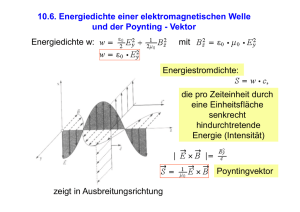
Polymere - familielindner.net
Werbung

1. Einleitung 1.1 Historische Entwicklung vor 1800 1839 1846 1868 1927 1931 1937 1943 1948 1959 1965 70er 80er Natürliche polymere Materialien wie Wolle, Seide, Horn, ... Vulkanisierung von Gummi Nitrierung von Zellulose Zelluloid Polyvinylchlorid PVC Polymethylmethacrylat PMMA Polystrol PS Teflon, Silicone und Polyurethane Kopolymere Thermoplasten Elastomere Styrol-butadien Blockkopolymere Thermothropische Flüssigkristalle Polyetheretherketon PEEK 1.2 Definitionen Monomere: kleine Moleküle, aus denen durch chemische Verknüpfung Polymere entstehen können, also Basisbausteine der Polymere (bestimmen chemische Eigenschaften von Polymeren) Oligomere: kurzkettige Polymere mit niedriger Molmasse (meist andere physikalische Eigenschaften) Polymerisation: Überführung der niedermolekularen Verbindungen (Monomere) in hochmolekulare Verbindungen (Polymere) Makromoleküle: Moleküle, in denen eine oder mehrere Arten von Atomen oder Atomgruppen zu langen Ketten oder verzweigten Netzwerken verknüpft sind (bestimmen physikalische Eigenschaften von Polymeren) Polymere: Makromoleküle, in denen eine Art oder mehrere Arten von Atomen oder Atomgruppen wiederholt aneinandergereiht sind, es gibt also Untereinheiten Polymerisationsgrad: Anzahl der Monomere, aus denen ein Polymer aufgebaut ist (da in der Praxis Polymere unterschiedlich lange Molekülketten enthalten, wird der Mittelwert angegeben) -1- Kopolymere bestehen aus zwei oder mehr verschiedenen Arten von Monomeren Konstitution: Die Konstitution eines Makromoleküls gibt Auskunft über die Art und die Anordnung der Grundbausteine und die dadurch bedingte Molekularstruktur. Beispiele (siehe auch Kopolymere): Homopolymer: ... AAAAAAAAAAA ... Alternierendes Kopolymer: ... ABABABABABA ... Konfiguration: beschreibt die räumliche Anordnung der Atome innerhalb des Moleküls Taktizität: Die Taktizität (Stereoregularität) beschreibt die in bestimmten Intervallen wiederkehrende Anordnung von Seitenketten in einem Polymer. Prinzipiell kann Taktizität nur bei Polymeren auftreten, die aus asymmetrischen Monomeren aufgebaut sind. Die Taktizität eines Polymers beeinflusst seinen räumlichen Aufbau. Je gleichmäßiger der Aufbau ist, desto leichter ist die Ausbildung einer Kristallstruktur. Konformation: Als verschiedene Konformationen eines Makromoleküls bezeichnet man die Konfigurationen, welche sich aus Rotationen um Bonds ergeben -2- 1.3 Polymerarchitektur (neben linear) 1.4 Verschiedene Polymere im Überblick -3- Polypropylen (PP): Polyvinylchlorid (PVC): Polyvinylidenfluorid (PVDF): Butadien-Kautschuk (BR): Naturkautschuk, Polyisopren (NR, IR): Polychloropren-Kautschuk (CR): Acrylnitril-Butadien-Kautschuk (NBR): Polyoxymethylen, Polyacetal (POM): Polyamid PA 6: Polyamid PA 66: -4- Styrol-Butadien-Kautschuk (SBR): Ethylen-Propylen-Kautschuk (EPM): Schlagfestes Polystyrol (SB): Handelsnamen: Polyethylen: Vestolen® A / EM, Hostalen® G, Lupolen® LD / HD Polypropylen: Vestolen® P, Hostylen® PP, Novolen® Polystyrol: Vestyron®, Polystyrol® Polyvinylchlorid: Vinoflex®, Vestolit® Polytetrafluorethylen: dyneon®, Teflon® Polyvinylidenfluorid: Dyflor® Polymethylmethacrylat: Lucryl®, Degalen®, Plexiglas® Butadien-Kautschuk: Buna CB Naturkautschuk, Polyisopren: Natsyn® 2000 Polychloropren-Kautschuk: Baypren®, Neoprene® Acrylnitril-Butadien-Kautschuk: Perbunan® Polyoxymethylen (Polyacetal): Delrin® PA 6: Perlon PA 66: Nylon, Ultramid® Styrol-Butadien-Kautschuk: Buna EM Ethylen-Propylen-Kautschuk: Buna AP Schlagfestes Polystyrol: Luran® -5- 2. Kettendimensionen Monodisperse Polymere: einheitliche Molekülgröße Polydisperse Polymere: Molmassenverteilung – mittleres Molekulargewicht 2.1 Mittelwerte über viele Ketten 2.1.1 Zahlenmittelwert MN = 1 N ∑N M i i = µ1 , mit N = ∑ N i Ni : Anzahl der Makromoleküle der Komponente i M i : Molmasse der Komponente i µ k : k-tes Moment Differentielle Form: M n = ∫ p ( M ) MdM p (M ) : Verteilungsfunktion 2.1.2 Gewichtsmittelwert MW ∑w M = ∑ wi i i ∑N M = ∑N M i 2 i i i = µ2 µ1 wi : Gesamtmasse der Makromoleküle der Komponente i Differentielle Form: M W = ∫ p( M )MMdM ∫ p(M ) MdM -6- 2.1.3 Polydispersivität U= MW MN −1 U : Uneinheitlichkeit (Maß für die Breite der Verteilungskurve, typisch zwischen 0,5 und 1) 2.1.4 Verteilungsfunktionen Hängen zumeist von der Synthesemethode ab: • Schulz-Zimm-Verteilung („step polymerization“): 1 β p( N ) = Γ( β ) N n β N β −1 exp − β N N n β : Wahrscheinlichkeit, dass Monomer angelagert wird U= • 1 β Poisson-Verteilung („chain polymerization“): ( p ( N ) = exp − N n U= ) Γ((NN +) 1) ≈ exp(− N ) NN e N n n n 1 Nn -7- N 2.2 Messmethoden: 2.3 Kettenkonformation Betrachtung: einzelne Polymerkette mit bestimmter Länge (keine Wechselwirkung mit anderen Ketten oder mit dem Lösungsmittel) 2.3.1 End-zu-End-Vektor v r : End-zu-End-Vektor („End-zu-End-Abstand“) v l i : Bindungsvektor („Bindungslänge“) Ai : Skelettatome v v r = ∑ li i N r 2 = ∑ li2 + 2 i =0 N vv ∑l l i j 0≤i < j ≤ N mit N : Polymerisationsgrad -8- vv li l j N ∑ i=0 0≤ i < j ≤ N Thermischer (statistischer) Mittelwert: Rl2 = r 2 = ∑ li2 + 2 vv Spezialfall gleicher Bindungslänge l (Homopolymer): Rl2 = Nl 2 + 2∑ li l j i< j 2.3.2 Trägheitsradius (Gyrationsradius) v si : Abstand(svektor) der Skelettatome Ai vom Schwerpunkt der Kette (Gyrationsradius) j v v rij2 = ∑ l k k =i s2 = 1 ( N + 1) 2 v2 ∑r ij 0≤i < j ≤ N Thermischer (statistischer) Mittelwert: R g2 = s 2 = 1 ( N + 1) 2 2.4 Spezielle Kettenmodelle 2.4.1 Frei verbundene Kette • v Bindungslänge fest und für alle Bindungen l i = l • Alle Bindungswinkel erlaubt • Alle Rotationswinkel erlaubt Maximale Kettenlänge: L = Nl End-zu-End-Abstand: Rl2 = Nl 2 → Rl ∝ N Gyrationsradius: R g2 = 1 2 Rl → Rl = N l 6 -9- ∑ i< j v rij2 Definition: Charakteristisches Verhältnis einer Kette (Polymerisationsgrad): c N = Rl2 Nl 2 =1 2.4.2 Frei rotierende Kette • v Bindungslänge fest und für alle Bindungen l i = l • • Bindungswinkel fest und für alle Bindungen θ i = θ Alle Rotationswinkel erlaubt End-zu-End-Abstand: Rl2 = Nl 2 1 + cos θ 1 − cos θ Charakteristisches Verhältnis: c N = 1 + cos θ 1 − cos θ Für N → ∞ gilt das Ergebnis der frei verbundenen Kette 2.4.3 Kette mit separierbarer Energie → • Sterische Behinderung wegen Seitengruppen • Rotationswinkel φ i nicht mehr frei wählbar mit dem im Bindungsrotationspotential festgelegtem Rotationswinkel φ i ist die Gesamtenergie der Kette für jede Konfiguration in Einzelbeiträge der Bindungen separierbar g+, g-: gauche+, gauche- 10 - • Nicht alle Rotationswinkel φ i sind gleich wahrscheinlich! • Unterschiedliche potentielle Energie (Minima und Maxima bei Drehung) • Unterschiedliche Wahrscheinlichkeiten für verschiedene Rotationswinkel → Berechnung durch statistische Mechanik End-zu-End-Abstand: Rl2 = Nl 2 1 + cos θ 1 + cos φ 1 − cos θ 1 − cos φ Charakteristisches Verhältnis (für N → ∞ ): c∞ = für N >>1, cos θ <1 1 + cos θ 1 + cos φ 1 − cos θ 1 − cos φ Beispiele: 2.4.4 Kette mit diskreten isomeren Rotationszuständen („RIS“ – Rotational Isomeric States) • Interdependez der Bindungsrotationspotentiale in der Polymerkette (Rotationswinkel am Knoten φ i nicht unabhängig vom Rotationswinkel an den Knoten φ i −1 und φ i +1 ) • Unterdrückung der gauche+, gauche- Konfiguration („Pentaneffekt“) 2.4.5 Kuhnsches Segment Um das einfache mathematische Konzept der frei beweglichen Kette an die Realität anzupassen, wird das Kuhnsche Segment eingeführt. Es besitzt eine effektive Segmentlänge l ' , ab der sich die Kette wie eine frei bewegliche Kette beschreiben lässt. Rl2 = N ' l ' 2 = c N Nl 2 , L = N 'l ' , L : Konturenlänge der Kette - 11 - 3. Mischungen, Lösungen, Schmelzen Polymerlösungen: Flory-Huggins-Theorie erklärt Eigenschaften von Polymermischungen und ermöglicht ein grundsätzliches Verständnis der Phasendiagramme. Beschreibung durch Änderung der freien Enthalpie: ∆Gm = ∆H m − T∆S m ∆H m : Änderung der Mischungsenthalpie T : Temperatur (=const., ebenso wie Druck p ) ∆S m : Änderung der Mischungsentropie: ∆S m = k B ln Ω k B : Boltzmann-Konstante Ω : Zahl der möglichen Anordnungen der Teilchen auf dem Gitter 3.1 Monomere („Ideale Gase“) ∆Gm = −T∆S m Gittermodell: ∆S m = −k B N (φ1 ln φ1 + φ 2 ln φ 2 ) • Keine Wechselwirkung zwischen den Teilchen • Keine Doppelbesetzung • Alle Gitterplätze sind besetzt ( N = N 1 + N 2 ) • N1 , N 2 : Teilchenzahl Sorte 1 bzw. Sorte 2 • φ1, 2 : Volumenbruch ( φ1, 2 = N 1, 2 / N ) ∆Gm ≤ 0 - 12 - 3.2 Polymer in Lösungsmittel (LM) Polymerisation der Teilchen der Sorte 2: • • • • • • • Keine Wechselwirkung zwischen den Teilchen Keine Doppelbesetzung Alle Gitterplätze sind besetzt ( N = N 1 + xN 2 ) N 1 : Anzahl der LM Moleküle N 2 : Anzahl der Polymerketten x : Zahl der Segmente pro Kette (Polymerisationsgrad) φ1, 2 : Volumenbruch • • z : Koordinationszahl vi +1 : Konfigurationsmöglichkeiten der i + 1 Kette φ ∆S m = − k B N φ1 ln φ1 + 2 ln φ 2 x mit φ1 = N1 xN 2 und φ 2 = N N • Lange Polymere sind schwer löslich (Entropiegewinn zu klein) • Mischungsentropie ist umso kleiner, je länger die Ketten sind • Polymere sind schlechter löslich als kleine Moleküle (Legierungen, Gase) 3.3 Mischung zweier Polymere Kettenlänge (Polymerisationsgrad) x1 und x 2 (Wechselwirkung zwischen den Polymeren): φ φ ∆S m = −k B N 1 ln φ1 + 2 ln φ 2 x2 x1 3.4 Thermodynamische Potentiale W : Energie V : Volumen N : Teilchenzahl S : Entropie T : Temperatur p : Druck µ : Chemisches Potential Innere Energie U ( S , V , N ) : dU = TdS − pdV + µdN T= ∂U ∂S V ,N , −p= ∂U ∂V S ,N , µ= ∂U ∂N V ,S - 13 - Freie Energie F (T , V , N ) : dF = dU − SdT = − SdT − pdV + µdN Enthalpie H ( S , p, N ) : dH = dU + pdV + Vdp = TdS + Vdp + µdN Freie Enthalpie („Gibbs free energy“) G (T , p, N ) : dG = − SdT + Vdp + µdN 3.5 Flory-Huggens-Theory 3.5.1 Flory-Huggens-Gleichung für „Polymer in LM“: φ ∆Gm = k B TN φ1 ln φ1 + 2 ln φ 2 + χφ1φ 2 x Flory-Huggens-Parameter χ = a +b k BT a , b : Wechselwirkungsparameter (experimentell) 3.5.2 Flory-Huggens-Gleichungen für Polymermischungen: φ φ ∆Gm = k B T 1 ln φ1 + 2 ln φ 2 + χφ1φ 2 x2 x1 3.6 Osmotischer Druck Reines LM p1 Polymerlösung p2 p1 < p 2 φ 1 Π = p 2 − p1 → Beschreibung des osmotischen Druckes: ΠV1 ≅ k B T 2 + − χ φ 22 x 2 • Hohe Temperaturen: χ ist klein → Wechselwirkung LM – Polymer ist gut (Polymere stoßen sich ab, sind ausgedehnt, „gutes“ LM) - 14 - • Tiefe Temperaturen: χ ist groß → Abstoßung LM – Polymer (Polymere ziehen sich an, Ketten ziehen sich zusammen, „schlechtes“ LM) • Θ -Temperatur: Aufhebung beider Effekte, ideale Konformation der Polymerketten, Polymer verhält sich wie im Vakuum χ (T = Θ) = 1 2 3.7 Phasenseparation Homogenes System aus LM und Polymer → System zerfällt in unterschiedliche Phasen z. B.: φα : hoher LM-Anteil φ β : niedriger LM-Anteil Stabilität der Mischung (Gibbs’sches Stabilitätskriterium): ∆G (φ ) < αG (φα ) + βG (φ β ) mit φ = αφα + βφ β und α + β = 1 → Berechnung von α und β ! 3.7.1 Phasendiagramm • Binodale: Trennlinie zwischen Ein- und Zweiphasengebiet - 15 - • Spinodale: Trennlinie zwischen metastabil entmischtem und stabil entmischtem Bereich Innerhalb der Spinodalen: spontane Entmischung Im metastabilen Bereich: mischung nach Keimbildung Ent- Tendenz zur Mischung ist bei Polymeren gegenüber der von niedermolekularen Systemen stark reduziert. Mischbarkeit findet man nur bei Systemen mit speziellen Monomerwechselwirkungen (Dipol-Dipol WW, Wasserstoffbrückenbindungen, Donor-Akzeptor WW). Speziell: Tieftemperaturentmischung (UCST) Hochtemperaturentmischung (LCST) - 16 - 4. Mechanische Eigenschaften Bindungskräfte steigen von: Van der Waals → Wasserstoffbrücken → kovalente Bindung Rückstellkräfte steigen von: Rotation um Bindungswinkel → Änderung des Bindungswinkels → Strecken (Komprimieren) der Bindungslänge Je mehr Polymerketten pro Fläche, desto stärker die Rückstellkräfte. 4.1 Definitionen σ xx F Spannung: σ = = σ yx A σ zx Dehnung: ε = σ xy σ xz σ yy σ yz σ zy σ zz ∆l l Hooksches Gesetz: σ = Eε ε = Dσ mit E : Young Modul (Elastizitätsmodul) mit D : Dehnnachgiebigkeit (tensile compliance) Tensordarstellung: Tij = cijkl S kl cijkl = λδ ij δ kl + µ1δ ik δ jl + µ 2δ il δ jl S ij = sijkl Tkl S ij = Querdehnung: ∆d = ε q = − µε d Scherung: γ = ∆x l τ = Gγ γ = Jτ 1 ∂u i ∂u j + 2 ∂x j ∂xi mit µ : Poisson-Zahl (Querdehnungszahl) mit G : Scher-, Schubmodul mit J : Schernachgiebigkeit Kompression: p = − K ∆V mit K : Kompressionsmodul (= 1 / χ mit χ : Kompressibilität) V Zusammenhang: E = 2(1 + µ )G = 3(1 − 2 µ ) K Komponenten Dehnungstensor: ε ik = 1 1 1 δ ik ∑ σ ii + σ ik − δ ik ∑ σ ii 9K 2G 3 i i - 17 - Torsion (Drillung): ϕ = 2l M πGr 4 mit G : Torsionsmodul = Schubmodul M : Drehmoment Viskoses Verhalten (Newton): τ = ηγ& mit η : Viskosität Jede Deformation kann als Summe einer reinen Scherung (Deformation ohne Volumenänderung) und einer homogenen Dilatation (Deformation mit Volumenänderung ohne Änderung der Form des Körpers) beschrieben werden. 4.2 Rheologische Modelle - 18 - 4.3 Temperaturabhängigkeit des E-Moduls 1: glasartig (hart, elastisch) 2: lederartig (elastischer + reversibler relaxierender Anteil) 3: gummiartig (elastisch, weich, reversibel); Gummielastizität: oberhalb der Glastemperatur → gestreckte Ketten ziehen sich wieder zusammen wegen Entropieerhöhung - umgekehrt wie bei Gasausdehnung. 4: viskos, irreversibel, Fließen, Bewegung der Ketten relativ zueinander 5: weich, viskos, vergleichbar mit flüssig Relaxationen im Glaszustand sind dominiert von lokaler Struktur (chemische Struktur der Monomere, Seitenketten, ...), Gummi- und viskoses Verhalten sind dominiert von globalen Eigenschaften des Polymers (Molekulargewicht, Molekulargewichtsverteilung, Vernetzungsgrad, ...). Temperaturabhängigkeit des mechanischen Relaxationsprozesses: Williams-Landel-Ferry (WLF) Gleichung (äquivalent zur Vogel-Fulcher-Tamann-Hesse (VFTH) Gleichung für dielektrische Relaxationsprozesse) log aT = − aT : C1 (T − Tr ) C2 + T − Tr Temperaturabhängiges Verhältnis der mechanischen Relaxationszeit τ (T ) zur Relaxationszeit τ 0 bei einer Referenztemperatur Tr . C1 , C 2 : WLF-Parameter - 19 - 4.4 Polymere unter mechanischer Belastung Spannungs-Dehnungskurven: Ideal plastisches Verhalten: Übergang elastisch → plastisch (also Übergang reversibler → irreversibler Verformung) am „yield point“. Unterschied viskoses - plastisches Verhalten: Temperatur niedrig: plastisches Verformen, Temperatur hoch: viskos; plastisches Fließen endet im Bruch! (a) spröder Bruch (b) duktiler Bruch (c) „necking“ Bruch (d) homogene Deformation Mikrorisse: Bei einer aufgezwungenen Deformation ist es möglich, dass nicht mehr ausreichend Zeit bleibt, damit sich alle Einheiten des Kunststoffes konform zueinander verformen. Diese Gefahr ist umso größer, je höher die Dehngeschwindigkeit und je inhomogener (Orientierung, Spurenelemente, ...) solche Stoffe sind. Bei einer kritischen Dehnung brechen gewisse Grenzflächen von Strukturelementen auf, wodurch Mikrorisse entstehen. Überschreiten die Mikrorisse eine gewisse Länge, so werden Spannungen in benachbarten Gebieten so groß, dass örtliche, irreversible Fließprozesse stattfinden. Je höher die Dehngeschwindigkeit, desto kleiner sind die Mikrorisse. In amorphen Kunststoffen werden diese Fließzonen auch Crazes genannt. Bei semikristallinen Materialien entstehen kleine, mit bloßem Auge nicht erkennbare Risse (Weißfärbung – „Weißbruch“). Für diese Kunststoffklasse ist der Bereich Tg < T < Tm am technisch wichtigsten, da dort das mechanische Verhalten durch das viskoelastische Verhalten des amorphen Anteils dominiert wird. - 20 - 4.5 Elastische Wellen Tα = cαβ S β ∂y dx0 ∂x0 ∂y S= = dx0 ∂x 0 dm y y+dx0·∂y/ ∂x0 x0 x0+dx0 T ∂2 y ∂2 y ∂T ∂T ∂2 y ∂T = A ρ dx = TA − T + dx A = − dx A → ρ =− 0 0 0 2 2 2 ∂x0 ∂x0 ∂x0 ∂t ∂t ∂t 2 ∂y ∂2 y c ∂2 y 2 ∂ y T = cS = c → 2 =− = −v →v= ∂x0 ρ ∂x02 ∂t ∂x02 ∂ 2u x ∂ 2u x ∂ 2u x ∂ 2u 2 + Allgemein: ρ 2 = c11 + c 44 ∂t ∂x 2 ∂z 2 ∂y Für Luft: c = c ρ ∂ 2u y ∂ 2u z + (c12 + c 44 ) + ∂x∂y ∂x∂z κp0 , κp 0 = Κ ρ (T ) - 21 - 5. Thermische Eigenschaften 5.1 Polymerzustände Die temperaturabhängigen Veränderungen der Struktur und der Eigenschaften von Polymeren erfolgen nicht kontinuierlich, sondern in Bereichen: Energieelastischer Zustand (hartelastischer Bereich): Im energieelastischen Bereich sind die Molekularbewegungen eingeschränkt, die Makromoleküle sind nicht frei gegeneinander verschiebbar, weshalb Kunststoffe in diesem Zustand prinzipiell hart und spröde sind (amorphe Thermoplaste, Elaste, Duroplaste). In teilkristallinen Thermoplasten sind die amorphen Bereiche besonders ungeordnet, wodurch hier eine gewisse Beweglichkeit und damit Zähigkeit ermöglicht wird. Die kristallinen Gebiete geben eine hohe Festigkeit. Typische Schubmodulwerte: 102 ÷ 104 N/mm2 Erweichungszustand (Einfrierbereich oder Glasübergangsbereich): Der Erweichungsbereich ist der Übergang vom energie- in den entropieelastischen Zustand. Die zwischenmolekularen Kräfte in den amorphen Bereichen werden zusehends durch Wärmebewegung überwunden, die Makromoleküle werden beweglich. Einen Erweichungsbereich - und somit eine Glastemperatur Tg - gibt es nur für amorphe Phasen. Bei teilkristallinen Thermoplasten besteht oberhalb der Erweichung des amorphen Bereiches ein Nebeneinander eines energieelastischen und eines entropieelastischen Zustandes. Es besteht daher für die kristallinen Bereiche bis zum Schmelzpunkt eine reine Energieelastizität ohne Glasübergang und entropieelastischen Bereich. Entropieelastischer Zustand (auch gummi- oder weichelastischer Bereich): Voraussetzung für ein entropieelastisches Verhalten ist die weitmaschige Verknüpfung der Makromoleküle. Die Beweglichkeit der Kettenabschnitte beruht auf der gegenseitigen Drehbarkeit der C-Atome um ihre Bindungsachse. Die Kettensegmente befinden sich wegen der Wärmebewegung im Zustand der größten Unordnung (= größte Entropie). Unter Zug weichen die Segmente dem äußeren Zwang aus, indem sie sich strecken (= Abnahme der Entropie). Mit Nachlassen der äußeren Kraft streben die Kettenteile wieder den Zustand größtmöglicher Unordnung an, wodurch die - 22 - ursprüngliche Gestalt erneut eingenommen wird („Memory-Effekt“: Strecken – Einfrieren – Erwärmen – ursprüngliche Gestalt). Bei Thermoplasten ist der entropieelastischen Verformung ein Fließen überlagert, das durch Abgleiten der Molekülketten wegen fehlender Vernetzung hervorgerufen wird. Eine vollständige Rückstellung der Dehnung bei Thermoplasten ist demnach nicht möglich (ev. Ausheilen durch Temperaturerhöhung). Schmelz-, Fließbereich: Oberhalb des entropie- oder gummielastischen Bereiches beginnt bei Thermoplasten der Übergang in den „plastischen“ Zustand der Schmelze (= Fließbereich), die Kettenmoleküle gehen in freie gegenseitige Beweglichkeit über (Kunststoffe verhalten sich im Zustand der „Schmelze“ zähflüssig, deshalb spricht man irreführenderweise von einem „plastischen“ Bereich). Die kristallinen Phasen besitzen einen engen Schmelzbereich. Bei teilkristallinen Kunststoffen wird ähnlich wie bei anderen kristallinen Werkstoffen ein eng begrenzter Schmelzbereich angegeben. 5.2 Klassifikation von Polymeren 5.2.1 Thermoplaste (Plastomere) Plastisch verformbare Polymere. Unvernetzte Produkte, deren Gebrauchstemperatur unterhalb der Schmelztemperatur und oberhalb der Glasumwandlungstemperatur Tg liegt. • Amorphe Thermoplaste (Gebrauchsbereich: 60 ÷ 80°C, Glastemperatur: variabel (durch „Weichmacher“ beeinflussbar, G-Modul: 102 ÷ 104 N/mm2, Beispiele: PS, PC, PMMA, SAN, ABS) - 23 - • Teilkristalline Thermoplaste (Gebrauchsbereich: Entiqueelastscher Bereich, Glastemperatur: ~50°C, G-Modul: 100 ÷ 5000 N/mm2, Beispiele: PE, PP, PA, POM, PTFE) 5.2.2 Thermoplastische Elaste (Thermoplastische Elastomere) Bei Raumtemperatur gummieleastisch verformbare Polymere, bei Verarbeitungstemperatur plastisch verformbar. Im Molekül unterschiedliche Segmente die diese unterschiedlichen Aufgaben erfüllen. 5.2.3 Elaste (Elastomere) Gummielestisch verformbare Polymere schwach vernetzte Produkte, deren Gebrauchstemperatur oberhalb der Glasumwandlungstemperatur Tg liegt. „Ideale“ GummiMaterialien! Gebrauchsbereich: Entropieelastischer Zustand Glastemperatur: -80 ÷ -10°C G-Modul: < 100 N/mm2 Vernetzung bei der Formgebung → Warmumformen und Schweißen danach nicht möglich - 24 - 5.2.4 Duroplaste (Duromere) Nur geringe Verformbarkeit möglich, dreidimensionale engmaschige Vernetzung ergibt eine starre intermolekulare Fixierung. Gebrauchsbereich: Glaszustand Glastemperatur: ~100°C G-Modul: 5 ÷ 15000 N/mm2 Beispiele: UP, EP, PF, SI, PU Vernetzung bei der Formgebung 5.3 Thermische Ausdehnung • Unterschied zwischen kubischem β und linearem Ausdehnungskoeffizienten α . • Bei orientierten Polymeren auch negative Ausdehnungskoeffizienten Kettenkontraktion senkrecht zur Faserrichtung möglich • Polymere im Gegensatz zu Metallen sehr große thermische Ausdehnungskoeffizienten durch Beispiele: Quarz: α = 1 ⋅ 10 −6 /K Eisen, Stahl: α = 10 ⋅ 10 −6 /K PS: α = 60 ÷ 80 ⋅ 10 −6 /K PMMA: α = 50 ÷ 110 ⋅ 10 −6 /K PE: α = 100 ÷ 250 ⋅ 10 −6 /K Probleme bei Verarbeitung von verschiedenen Materialien (Polymer/Metall), die großem Temperaturbereich ausgesetzt sind (Auto, Flugzeug, Computer) → 5.4 Wärmekapazität • Wärmekapazität von Polymeren kleiner als die von Gasatomen, da immer einige Freiheitsgrade eingefroren sind Beispiele: Quarz: cV = 0.72 J/gK PVC: cV = 0.85 J/gK PS: cV = 1.30 J/gK PE: cV = 2.1 J/gK Wasser: cV = 4.2 J/gK - 25 - • T niedrig: lineares Anwachsen von cV , da Atome stärker um Ruheposition vibrieren • Bei Tg : starkes Anwachsen von cV , da zusätzlich Bewegung um Bindungen der Kette • Bei Tm : Maximum von cV , da Wärme zum Schmelzen der Kristalle benötigt wird (kleine kristalline Bereich schmelzen zuerst) • Unterschied zwischen amorphen (○) und semikristallinen (●) Polymeren: • Allgemein: T− Bereich Kristalline Polymere ( cV ∝ ) Art der Schwingung Schwingungsausbreitung I T3 (Debye) T 3 + T 3/ 2 (Debye + Stockmayer-Hecht) T + T 1/ 2 (Tarasov + Stockmayer-Hecht) Const. (Dulong-Petit) Streck 3D Streck Biege Streck Biege 3D II III IV 1D T− Bereich Amorphe Polymere ( cV ∝ ) Art der Schwingung Schwingungsausbreitung I T +T3 (Tunneling + Debye) T3 (Debye) T 3 + T 3/ 2 (Debye + Stockmayer-Hecht) T 0.8 , T 0.9 Streck 3D Streck 3D Streck Biege Streck Biege 3D II III IV V Const. (Dulong-Petit) - 26 - 1D 5.5 Wärmeleitfähigkeit • Polymere sind schlechte elektrische Leiter, da die Phononen freie Wellenlängen von 0.7 nm haben → Wärmeleitfähigkeit λ in Polymeren sehr viel kleiner als in Metallen. Beispiele: Quarz: λ = 10.5 W/mK Metall: λ = 100 W/mK PS: λ = 0.16 W/mK PE: λ = 0.35 W/mK • Wärmeleitfähigkeit ist für geordnete, semikristalline Systeme besser als für ungeordnete, amorphe Systeme. • Kaum Änderung der Wärmeleitfähigkeit bei Tg , da sich die Packungsdichte der Segmente nicht deutlich ändert; mit steigender Temperatur sinkt die Packungsdichte der Segmente → λ nimmt ab. • Wärmeleitung: j v = −λ 1 3 dT dx mit l : mittlere freie Weglänge λ = cV ρvl o Hohe Temperaturen – Streuung an 1 1 N ∝ T ∨ ∝ N → l ∝ → λ ∝ (Peierls Umklapp-Prozesse) T T τ Phononen: 1 o Tiefe Temperaturen – Streuung an Kristallfehlern: λ ≈ cV vD → λ ∝ T 3 5.6 Definitionen Entropie: S = − Volumen: ∂G ∂T p ∂G ∂p T Enthalpie: H = G + TS = G − T ∂G ∂T p 2 Wärmekapazität: cV , p = −T ∂ G ∂T 2 V , p cV = Kubischer Ausdehnungskoeffizient: β = Kubische Kompressibilität: κ = − 1 ∂V V ∂p 1 ∂V V ∂T ∂U ∂T p T - 27 - V 5.7 Experimentelle Methoden 5.7.1 Differential Scanning Calorimetry (DSC) • Messung der Wärmekapazität • Probe und Referenzmaterial auf gleicher Temperatur • Messung der elektrischen Leistung, die notwendig sind, um kalorische Effekte zu kompensieren • Messung der Temperaturabhängigkeit der zugeführten Wärme Messung der Fest-Fest-Übergangstemperatur TSS , Glasübergangstemperatur TG , Flüssig-Flüssig-Übergangstemperatur Tll , Kristallisationstemperatur Tcryst , kristallinen Schmelztemperatur TM , exothermen Reaktionstemperatur Treact und Zersetzungstemperatur Tdecomp . 5.7.2 Thermogravimetrie (TGA) • Messung der Probenzusammensetzung • Messung des Gewichtsverlustes beim Aufheizen der Probe 5.7.3 Differential Thermal Analysis (DTA) • Messung der Wärmekapazität • Probe und Referenzmaterial mit konstanter Temperaturrate geheizt • Beobachtung der Temperaturdifferenz zwischen Probe und Referenzmaterial (unterschiedlicher Wärmefluss) - 28 - 5.7.4 Thermo Mechanical Analysis (TMA) • Messung des Ausdehnungskoeffizienten • Mechanische Antwort als Funktion der Temperatur Details zur Glasübergangstemperatur Tg → Phasenübergänge - 29 - 6. Elektrische und Dielektrische Eigenschaften Die wichtigsten elektrischen Eigenschaften von Kunststoffen sind: • • • • • • • hoher spezifischer Widerstand relativ hohe Dielektrizitätszahl unterschiedliche dielektrische Verlustfaktoren gute Durchschlagsfestigkeit Kriechstromfestigkeit Lichtbogenfestigkeit guter Oberflächenwiderstand 6.1 Elektrische Leitfähigkeit Für die Leitfähigkeit sind Transportvorgänge von Ladungsträgern notwendig. Bei den Ladungsträgern kann es sich um Elektronen oder Ionen handeln. Da in Kunststoffen höchstens durch Verunreinigungen freie Elektronen vorhanden sind, sind Ionen für die Leitung verantwortlich. Dabei sind insbesondere Weichmachermoleküle zu nennen, die sich zwischen die Ketten einlagern und Stromtransporte erleichtern. Ebenso wirken auch Wassermoleküle. Der elektrische Widerstand nimmt mit zunehmender Temperatur ab, da die Moleküle beweglicher werden. Beispiele: PTFE: 10-16 S/m Polyacetylen (undotiert): 10-10 S/m Silber, Kupfer: 108 S/m Bestimmte Polymere → bereits bei Raumtemperatur geringe el. Leitfähigkeit → Peierls Übergang: Bändermodell für eine unendlich ausgedehnte Kette von Atomen im gleichen Abstand: Band zur Hälfte mit Elektronen gefüllt; Günstiger (energieärmer): jeweils zwei Atome sitzen dichter beieinander (Peierls-Verzerrung) → Band teilt sich in ein besetztes Band (Valenzband) und ein unbesetztes Band (Leitungsband) auf, das Material ist halbleitend. - 30 - Wechsel zwischen Einzel- und Doppelbindungen ohne Energieänderung, Bildung von freien Radikalen dort, wo Zustand A und B zusammentreffen (Defekt = neutrales Soliton) → Ladung des freien Elektrons wird über alle Monomereinheiten verteilt Die Peierls-Verzerrung kann aufgehoben werden, wenn durch Dotierung Elektronen entfernt oder zugefügt werden, das Polymer wird metallisch leitend! Beispiele: stabile Solitonen (entarteter Grundzustand) instabile Solitonen (nichtentarteter Grundzustand) 6.2 Dotieren Vergleich: Polyacetylen (undotiert): 10-10 S/m Polyacetylen (dotiert): 105 S/m • Bedingung: abwechselnde Folge von Einfach- und Doppelbindungen • Dotieren durch Oxidation (Akzeptorzustand) oder Reduktion (Donorzustand) → Doppelbindung delokalisiert • Zusammenführung von Valenz- und Leitungsband → metallische Leitfähigkeit 6.3 Photoleitfähige Polymere In Isolatoren kann durch Lichteinstrahlung ein Strom in einem elektrischen Feld erzeugt werden. Lichteinstrahlung produziert also freie Ladungsträger. • Elektronen, die lose an eine Anionenleerstelle gebunden sind, absorbieren Licht • Lose gebundenes Elektron wird ins Leitungsband angeregt • Dort bewegt es sich so lange bis es wieder eingefangen wird • Gemessener Strom entspricht der Verschiebung der Elektronen im elektrischen Feld. - 31 - 6.3.1 Beispiele • Polymere mit Seitengruppen: Leitfähigkeit in den Seitengruppen (z. B. Poly-N-vinyl-carbazol (PVK)) • Konjugierte Polymere: Einfachbindung zwischen zwei Doppelbindungen; Ein konjugiertes π − System entsteht aus der Kombination von p − Elektronenorbitalen zu über mehreren Atomen delokalisierten Molekülorbitalen. Konjugierte Polymere sind dadurch gekennzeichnet, dass das π − Elektronensystem erst bei der Synthese gebildet wird. Eine besondere Eigenschaft ist die allgemein schlechte Löslichkeit in Lösungsmitteln. Aufgrund des relativ geringen HOMO-LUMO-Abstandes sind solche Polymere oft gefärbt (Farbzentren, „traps“) oder haben Absorptionen im UV-A Bereich (HOMO: höchstes besetztes Molekülorbital, LUMO: niedrigstes unbesetztes Molekülorbital; eine elektronische Anregung kann als Anheben eines Elektrons aus dem HOMO ins LUMO aufgefasst werden, eine Oxidation als das Entfernen eines Elektrons aus dem HOMO und eine Reduktion als das Hinzufügen eines Elektrons ins LUMO) (z. B. Polyacetylen (PA), Poly(phenylenvinylen) (PPV), Polyphenylen, Polythiopen, Polypyrrol, Polyanilin, Polysilan) 6.3.2 Mechanismus: Excitonen • Einfallendes Licht hebt Elektron aus dem Valenzband ins freie Leitungsband → Schaffung eines Elektron-Loch Paares (Picosekunden-Bereich); Bandabstand ca. 4 eV • Schwingungsanregung des Elektron-Loch-Zustandes → Exciton • Van-der-Waals WW → geringe Überlappung der Wellenfunktionen • Ladungsträger über das ganze Gitter verteilt • Bewegung der Ladungsträger → Hüpfprozess! • Theoretische Beschreibung: - Beschreibung der Beweglichkeit (Onsager Ansatz) - Beschreibung der Beweglichkeit (Bäßler Modell) 1 ln µ ∝ − 2 ln µ ∝ E T - 32 - 6.3.3 Anwendungen Beschichtung für fotografische Filme, Strahlenschutzbeschichtungen, Selbsttönende Fenster, Elektrofotografie (Xerokopie): 6.4 Elektrolumineszenz Als Lumineszenz bezeichnet man - einfach ausgedrückt - die Eigenschaft bestimmter Substanzen, die in Folge von Bestrahlung mit Tageslicht, UV-, Röntgen- oder Elektronenstrahlen absorbierte Energie ganz oder teilweise zu emittieren. Die direkte Lumineszenzanregung durch die alleinige Einwirkung eines elektrischen Feldes nennt man Elektrolumineszenz. Hierzu werden fluoreszierende Stoffe bzw. Leuchtpigmente, Luminophore oder Phosphorverbindungen verwendet. In der Natur gibt es viele Beispiele für Lumineszenzeffekte. Der bekannteste ist wohl das Glühwürmchen, das sein gelbliches Licht ein- und ausschalten kann. Forscher haben die dahinter stehenden Grundlagen analysiert und festgestellt, dass einige natürliche Polymere Halbleitereigenschaften haben und somit für den Transport elektrischer Ladungen geeignet sind. Solche konjugierte Polymere können mittlerweile künstlich und genau spezifiziert hergestellt werden. Halbleiter und andere elektrische Bauteile sind also nicht mehr auf Kristallstrukturen angewiesen, sondern können aus Kunststoffen gefertigt werden. Es lassen sich aber auch andere, von kristallinen Halbleitern bekannte Effekte mit konjugierten Polymeren erzielen. Der Leuchteffekt ist hier die Parallele zur Lichtemission der seit Jahrzehnten genutzten Leuchtdioden. - 33 - Organische Leuchtdioden bestehen aus einer oder mehreren halbleitenden organischen Schichten, die zwischen zwei Elektroden, je einer Anode und Kathode, positioniert sind. Wird eine elektrische Spannung an die Elektroden gelegt, fließt Strom durch die organische Substanz und löst eine Licht-Immission aus. OLED-Elemente sind nicht dicker als 200 Nanometer. Durch chemisch unterschiedliche Schichten lassen sich gewünschte Farben erzeugen. 6.4.1 Definition, Aufbau • Elektrolumineszenz-Folien: Betrieb mit Wechselspannung (400÷800 Hz) • OLED-Folien: Betrieb mit Gleichspannung (vgl. Halbleiter LEDs) 6.4.2 Mechanismus bei entartetem Grundzustand: Excitonen • Strom-Spannungskennlinie → Schottky-(Halbleiter)Dioden Charakter • Elektron und Loch werden von den beiden Seiten des Polymers emittiert und rekombinieren im Inneren (= Excitonen-Erzeugung, welche durch Lumineszenz abregen). Der viel häufiger vorkommende Konkurrenzprozess (99%) ist strahlungslose Rekombination in Fehlstellen. • Beispiel: Poly-para-(phenylenvinylen) (PPV) (gelb-grüner Bereich) 6.4.3 Mechanismus bei nicht-entartetem Grundzustand: Polaronen • keine Soliton-Defekte die zur Leitfähigkeit führen • Lokalisierte Elektron-Lochpaare polarisieren lokal die Umgebung → Relaxation in neuen Grundzustand: Polaron (Kombination aus einem geladenen und neutralen Soliton, „Polarisationswelle“) • Polaron kann nur durch Überwinden der Energiebarriere bewegt werden • Kombination zweier geladenen Solitonen (Paar von Polaronen) → Bipolaron - 34 - • Bewegung der Ladungsträger → Hüpfprozess! • Beschreibung als Schottky-Diode inadequat • Beispiel: Poly-para-phenylen (PPP), Bandgap etwa 2.7 eV, blauer Bereich Im Gegensatz zu herkömmlichen LCD/TFT Elementen, bei denen eine auf der DisplayRückseite angeordnete Lichtquelle (Backlight) ständig in Betrieb ist, deren Lichtaustritt durch Steuerung der TFT Elemente reguliert wird, leuchten OLEDs eigenständig und ohne jede zusätzliche Lichtquelle. Vorteile OLEDs: Geringerer Energieverbrauch, schnellere Reaktionszeit, höhere Helligkeit und Kontrast, hervorragende Schwarzwerte, geringe Dicke und Gewicht, hohe Flexibilität und Widerstandsfähigkeit gegenüber Umgebungseinflüssen. 6.5 Durchschlagsfeldstärke (Durchschlagsfestigkeit) Der äußerst geringe Strom durch einen Isolierstoff nimmt im Bereich kleiner Spannungen proportional zur angelegten Spannung zu (Ohmsches Gesetz). Dann erfolgt bei weiterer Spannungszunahme eine überproportionale Erhöhung des Stromes und schließlich ein sprunghafter Anstieg. Es hat ein Durchschlag stattgefunden. Die Spannung, bei der der Durchschlag erfolgt, heißt Durchschlagspannung, die entsprechende Feldstärke heißt Durchschlagsfeldstärke E D . E D kann für ein und dieselbe Materialart wegen lokaler Inhomogenitäten und äußerer Einflüsse (mechanische Spannungen, chemische Einwirkungen) erheblich variieren. ED ∝ 1 T und ED = 1 f - 35 - Der Durchschlag erfolgt umso eher, je größer die Materialtemperatur und je höher die Frequenz bei elektrischen Wechselfeldern ist. 6.5.1 Durchschlagsmechanismen • Lawinendurchschlag (Erzeugung einer Elektronenlawine bei hohen elektrischen Feldern) • Elektromechanischer Durchschlag (Kurzschluss zwischen den Elektroden durch elektrostatische Anziehung) • Thermischer Durchschlag (Verstärkung von Restströmen bei erhöhten Temperaturen) • Innerer Durchschlag (durch Lufteinschlüsse verursachte Teilentladungen, die zum Durchschlag führen. Teilentladungen beginnen bei der Einsatzfeldstärke.) 6.6 Dielektrisches Verhalten von Polymeren - Definitionen Durch die Wirkung eines äußeren elektrischen Feldes werden die örtlichen Verteilungen der positiven und negativen Ladungen im Innern eines Isolators gegeneinander verschoben. Die dadurch entstehende elektrische Polarisation ist gekennzeichnet durch die Bildung von elektrischen Dipolen. • Passive Dielektrika: Elektrische Isolierstoffe • Aktive Dielektrika: Ausnutzung der Polarisationserscheinungen r v v v Für elektrisch homogene, isotrope und kubische Materialien gilt: D = εE ( D und E parallel) v D : Dielektrische Verschiebung, elektrische Flussdichte, Flächenladungsdichte ε : Permittivität, Dielektrizitätskonstante v In anisotropen v Materialien ist ε jedoch ein Tensor (richtungsabhängig) → D nicht unbedingt parallel zu E . v v Für elektrisch inhomogene Materialien gilt: ε = ε 0ε r → D = ε 0ε r E ε 0 = 8.854 ⋅ 10 −12 As / Vm : Permittivität des Vakuums, elektrische Feldkonstante ε r : relative Permittivität, Permittivitätszahl; Materialkenngröße (dimensionslos, temperaturabhängig, frequenzabhängig) Freie Ladung: Q frei A = D = ε0E - 36 - Gebundene (Polarisations-)Ladung: Q Pol = P → D = ε0E + P A Polarisation: P = ε 0 (ε r − 1) E = ε 0 χE mit χ : elektrische Suszeptibilität 6.7 Dielektrisches Verhalten von Polymeren – Frequenzabhängigkeit der Polarisation Gegen die Ladungsverschiebung bzw. das Ausrichten der Dipole im elektrischen Feld in Feldrichtung wirken Reibungskräfte. Dadurch entstehen im elektrischen Wechselfeld, d. h. bei periodischer Bewegung der Ladungen bzw. Dipole, Verluste. Als Folge dieser dielektrischen Verluste beträgt die Phasenverschiebung zwischen Strom und Spannung einer realen Kondensatoranordnung bei Speisung mit Wechselstrom nicht 90° sondern 90°- δ . Dabei ist δ der Verlustwinkel (ein Maß für die Dämpfung oder den Verlust) und führt zur Aufheizung. Darstellung durch den imaginären Anteil der Dielektrizitätskonstante: ε (ω ) = ε ' (ω ) − iε ' ' (ω ) tan δ = ε ' ' (ω ) ε ' (ω ) Die im Material absorbierte Energie wird zum größten Teil zur Polarisierung des Dielektrikums aufgewendet. Da der spezifische Widerstand nicht unendlich groß ist, treten außerdem geringe Leitungsverluste auf. Die dielektrischen Verluste verhalten sich analog zu den Ohmschen Wärmeverlusten in Leiterwerkstoffe. Man kann deshalb einen Kondensator mit Dielektrikum im Ersatzschaltbild durch einen idealen, verlustfreien Kondensator mit parallel geschaltetem Ohmschen Widerstand darstellen. 6.7.1 Polarisationskurve Bei höheren Frequenzen können die ionische und die Orientierungspolarisation dem anregenden elektrischen Feld nicht mehr folgen, übrig bleibt daher nur der Betrag der elektronischen Polarisation. - 37 - 6.7.2 Elektronenpolarisation v Ein elektrisches Feld E trennt den positiven Ladungsschwerpunkt (Atomkern) vom negativen Ladungsschwerpunkt (Zentrum der Elektronenhülle) der Atome. Es tritt eine Deformation der Elektronenhülle auf - die Atome werden polarisiert. Mit dieser Polarisation ist bei Festkörpern eine mechanische Längenänderung verbunden, die unabhängig von der Feldrichtung je nach Kristallrichtung positiv oder negativ sein kann. Man nennt diese Erscheinung Elektrostriktion. εM ∝ E2 mit ε= ∆l l Die Elektronenpolarisation tritt bei allen, die Elektrostriktion bei allen festen dielektrischen Werkstoffen auf. Die Umkehrung der Elektrostiktion, d. h. die Erzeugung einer Polarisation durch mechanische Verformung ist dagegen nur bei einer bestimmten Gruppe von dielektrischen Werkstoffen (Piezoelektrika) möglich. Die Elektronenpolarisation folgt wegen der geringen Masse einem elektrischen Wechselfeld bis zu Frequenzen von ungefähr 1015 Hz (UV-Bereich). Es gilt: ε r = n 2 mit n : optischer Brechungsindex Die Elektronenpolarisation ist temperaturunabhängig! Beispiele: Diamant, Ge, Si, PS, PTFE, PE Für die makroskopische Beschreibung der Elektronenpolarisation in unpolaren Festkörpern gilt das Clausius-Mosotti-Gesetz: χ χ +3 = ε r − 1 Nα = ε r + 2 3ε 0 mit α : mikroskopische (atomare) Polarisierbarkeit 6.7.3 Ionenpolarisation Sie tritt nur in Ionenkristallen auf. Das elektrische Feld verschiebt die Ladungsschwerpunkte positiver und negativer Ionen gegeneinander (Verschiebung des positiven Untergitters gegenüber dem negativen). - 38 - Die Ionenpolarisation ist temperaturunabhängig. Sie folgt einem elektrischen Wechselfeld aufgrund der größeren Masse der Ionen im Vergleich zu den Elektronen nur bis zu Frequenzen von 1013 Hz (Infrarot). Beispiele: Ionenkristalle, Keramiken, Gläser, NaCl 6.7.4 Orientierungspolarisation Sie tritt nur in Materialien mit bereits bei E = 0 vorhandenen elektrischen Dipolen (polare Materialien) auf. Das elektrische Feld übt auf die infolge der Temperatur statisch im Raume angeordneten elektrischen Dipole ein mechanisches Drehmoment aus und verursacht hierdurch eine teilweise Ausrichtung in Feldrichtung. Die Orientierungspolarisation ist stark temperaturabhängig. Mit höherer Temperatur nimmt sie ab. Eine Temperaturerhöhung wirkt dem ordnenden Einfluss des elektrischen Feldes entgegen. Die Orientierungspolarisation folgt einem elektrischen Wechselfeld nur bis zu der so genannten Relaxationsfrequenz, die sehr stark temperaturabhängig ist und Werte zwischen 106 bis 109 Hz annehmen kann (Mikrowellen bis UHF). Beispiele (polare Kunststoffe): PVC, PET, Kunstharz, Papier Hingegen: Unpolare Kunststoffe (keine Orientierungspolarisation): PE, PP, PTFE Die Abhängigkeit der dielektrischen Größen ε r und χ von der Feldfrequenz bezeichnet man als Dispersion oder als Dielektrisches Spektrum. Mit steigender Frequenz fällt zuerst die Orientierungs-, dann die Ionen- und schließlich die Elektronenpolarisation aus, da die einzelnen Polarisationsmechanismen der hohen Änderungsgeschwindigkeit des Feldes nicht mehr folgen können. Im Zusammenhang mit dieser Tatsache spricht man bei der Orientierungspolarisation von Relaxation, bei die Ionen- und Elektronenpolarisation von Resonanz. 6.8 Dielektrische Relaxionsprozesse 6.8.1 Dielektrische Relaxationsstärke Unterschied zischen den Permittivitäten im Glas- und Gummizustand → Kirkwood-FröhlichGleichung: ∆ε = ε r − ε u = 3ε r 2ε r + ε u 2 2 ε u + 2 gNµ 0 3 3ε 0 kT - 39 - ε r , ε u : Permittivitäten bei hoher/niedriger Frequenz/Temperatur µ 0 : Dipolmoment g : Korrelationsfunktion 6.8.2 Temperatur- , Frequenzabhängigkeit ε = ε '−iε ' ' = ε u + • ∆ε [1 + (iωτ (T )) ] p q mit Oberhalb Tg : α-Relaxation - Vogel-Fulcher-Tamann-Hesse (VFTH) Gleichung: B mit T − T 2 Normierung mit WLF-Gleichung τ (T ) = A exp • T2 : statische Glastemperatur Um Tg : Addam-Gibbs-Theorie: B T (1 − T2 / T f τ (T ) = A exp • τ (T ) : Mittlere Relaxationszeit ) mit T f : fiktive Temperatur Unterhalb Tg : β-Relaxation - Arrhenius-Verhalten Phänomenologisch lassen sich Relaxationsprozesse mit der Kohlrausch-Williams-Watts (KWW) Funktion beschreiben (anpassen): t φ (t ) = exp − τ (T ) p - 40 - 6.8.3 α-Relaxation (Hauptrelaxation) • Bewegung auf „großer Skala“ • Bei niedrigen Frequenzen • Mit Glasübergang verknüpft → kollektive Umlagerung der Polymerhauptketten • Modellbeschreibungen: Platzwechselmodell (Segmente bewegen sich ins freie Volumen), Rouse-Modell (Änderung der Konformation), Zimm-Modell (Hydrodynamische Effekte berücksichtigt), Reptationsmodell (Kriechen durch „selbstgeformte“ Röhren) • Sehr gute Beschreibung durch KWW-Funktion 6.8.4 β-Relaxation (Sekundärrelaxation) • Lokalisierte Bewegungen • Bei hohen Frequenzen • Bei hohen Temperaturen → α-Prozess • Durch Seitenarmbewegungen, lokale Bewegungen oder partielle Reorientierung der Hauptkette • Verschiebung der Anregungsmaxima zu höheren Frequenzen → γ-, δ-Relaxation (Sekundärrelaxationen) α- und β-Relaxationen sehr unterschiedlich für verschiedene Polymere, Verschmelzung oberhalb bestimmter Temperaturen („Cross-Over“) 6.9 Symmetrieeigenschaften • Allgemein: ε ij = α ik α lj ε kl • ε 11 Monoklines System: ε ij = α α ε ij , ε ij = ε 12 0 • i i j j ε 11 0 Orthorhombisches System: ε ij = 0 ε 22 0 0 - 41 - ε 12 ε 22 0 0 0 ε 33 0 0 ε 33 • 0 ε 11 0 Trigonales, Tetragonales System: ε ij = 0 ε 11 0 0 0 ε 33 • 0 ε 11 0 Kubisches System: ε ij = 0 ε 11 0 0 0 ε 11 6.10 Frequenzeigenschaften (Resonanzen) S1 = s11T1 − µs11T2 + d 31 E3 S 2 = s11T2 − µs11T1 + d 31 E3 D3 = d 31 (T1 + T2 ) + ε 33 E3 • Niedrige Frequenzen: T1 = T2 = 0 → D3 = ε 33 E3 • Mittlere Frequenzen: S1 = 0 ∧ T2 = 0 → D3 = ε 33 (1 − k 312 ) E3 , k 312 = d 312 s11ε 33 k 31 : elektromechanischer Kopplungsfaktor ≤ 1 • 2d 312 E3 Hohe Frequenzen: S1 = S 2 = 0 → D3 = ε 33 1 − ( 1 − µ ) s ε 11 33 Frequenz (Längenresonanzen) (Dickenresonanzen) < Frequenz - 42 - (Breitenresonanzen) < Frequenz 7. Lineare und Nichtlineare optische (NLO) Eigenschaften Im Gegensatz zur linearen (oder klassischen) Optik, sind die optischen Eigenschaften eines Materials, z. B. der Brechungsindex, nicht mehr nur eine Funktion der Wellenlänge des verwendeten Lichts und damit quasi Materialkonstanten. Bei großen Feldstärken sind sie zusätzlich von der Intensität des einfallenden Lichts abhängig. Die nichtlineare Optik befasst sich also mit optischen Materieeigenschaften, die durch das einwirkende Licht selbst verändert werden. Während konventionelle Lichtquellen keine messbaren nichtlinear optischen Effekte hervorrufen können, hat sich kurz nach der Erfindung des Lasers im Jahre 1960 und parallel mit dessen Fortentwicklung die Forschung rasch auf das Gebiet der nichtlinearen Optik ausgedehnt. Erst der Laser hat, in seiner Eigenschaft als Quelle für intensives und hochkohärentes Licht, die experimentelle Untersuchung der nichtlinearoptischen Eigenschaften geeigneter Materialien möglich gemacht und den Weg bereitet, deren Potential für zahlreiche neue Anwendungen in der optischen Signalverarbeitung zu erschließen. 7.1 Tensoren mit ei = Aij c j Aij = α ik α lj Akl Aij : physikalische Eigenschaften des Materials Skalar: Tensor 0. Stufe (z. B. Spezifische Wärme) Vektor: Tensor 1. Stufe (z. B. Pyroelektrizität) Matrix: Tensor 2. Stufe (z. B. Permittivität, Leitfähigkeit, Thermische Ausdehnung) Matrix: Tensor 3. Stufe (z. B. Piezoelektrizität, Pockels-Effekt, NLO-Effekte 2. Ordnung) Matrix: Tensor 4. Stufe (z. B. Elastizität, Elektrostriktion, Kerr-Effekt, NLOEffekte 3. Ordnung) Durch Wahl geeigneter Symmetrieeigenschaften → „Zusammenfassen“ von Indizes Allgemein gilt: Die Struktur bestimmt die Symmetrie, die Symmetrie bestimmt die Materialeigenschaften! 7.2 Dielektrische Funktion, Polarisation 7.2.1 Makroskopische Beschreibung ( ) ( 3) Di = Pi + ε 0 ε ij(1) E j + ε ijk( 2) E j E k + ε ijkl E j E k El + ... ( ) ( 3) Pi = ε 0 χ ij(1) E j + χ ijk( 2) E j E k + χ ijkl E j E k El + ... Lineare Optik NLO - 43 - mit Pi : eingefrorene Polarisation 7.2.2 Mikroskopische Beschreibung Molekulares Dipolmoment: µ i = αE , α : molekulare (lineare) Polarisierbarkeit Molekulare Polarisation: pi = µ i + ε 0 (α ij E j + β ijk E j E k + γ ijkl E j E k El + ...) β ijk , γ ijkl : Hyperpolarisierbarkeiten 1. und 2. Ordnung Effizienz NLO-Effekte 2. Ordnung hängt von 1. Hyperpolarisierbarkeit (molekularer Suszeptibilität 2. Ordnung) β ab → Beschreibung durch 2-Niveau-Modell bei amorphen NLO Polymeren: β (−ω 3 ; ω1 , ω 2 ) = e 3 µ 02 ∆µ ω 02 (3ω 02 + ω1ω 2 − ω 32 ) h2 (ω 02 − ω12 )(ω 02 − ω 22 )(ω 02 − ω 32 ) 7.2.3 Lineare Optik • Doppelbrechung (lineare optische Anisotropie nach Polung): ∆n = ε 0 N /V 2n 2 2 ε u + 2 µ0 F ∆α 3 45kT mit F : lokales Polungsfeld σ dc σ~ • Drude-Modell: ε~ = , σ~ = iωε 0 1 − iωτ Hagen-Rubens (leitender Bereich): ωτ << 1 → σ~ = σ dc σ σ Lorenz (Plasmabereich): ωτ >> 1 → σ~ = − dc , ε~ = 2 dc iωτ ω τε 0 Wellenwiderstand: R = 1 − 2 1 = n σd 7.2.4 NLO-Effekte 2. Ordnung (quadratische NLO-Effekte) Frequenzverdopplung (SHG – Second Harmonic Generation), linearer Pockels-Effekt, linearer Stark-Effekt (Elektroabsorption), Frequenzmischung, Faraday-Effekt, Optische Gleichrichtung Voraussetzung: Nicht-Zentrosymmetrie (d. h. kein Inversionszentrum): ( ) Inversion: − P = ε 0 − χ (1) E + χ ( 2 ) E 2 − χ ( 3) E 3 + ... χ ( 2) E 2 = 0 ( ) Gleichzeitig: − P = ε 0 − χ (1) E − χ ( 2) E 2 − χ ( 3) E 3 + ... - 44 - Wechselwirkung mit elektromagnetischer Welle: E = E 0 cos(ωt − kz ) → 1. NLO Term → • ( 2) (2ω ) = Frequenzverdopplung (SHG): PNL 1 ( 2) 2 χ E0 cos(2ωt − 2kz ) 2 χ ( 2) (−2ω ; ω , ω ) D ω, kω ω, kω - e Fundamentale 2ω, 2kω SHG A • ( 2) Optische Gleichrichtung: PNL ( 0) = 1 ( 2) 2 χ E0 2 χ ( 2) (0; ω ,−ω ) • Linearer elektrooptischer Effekt (EO Effekt) → Pockelseffekt: n ∝ E χ ( 2) (−ω ; ω ,0) • Differenz-Frequenzmischung (DFG): χ ( 2) (ω1 − ω 2 ; ω1 ,−ω 2 ) • Summen-Frequenzmischung: χ ( 2) (ω1 + ω 2 ; ω1 , ω 2 ) • Parametrischer Prozess: Zerfall eines Pump-Photons in zwei Photonen (z. B. zur Erzeugung von abstimmbarem Licht) (a) Frequenzverdopplung, (b) Frequenzmischung, (c) parametrischer Prozess, (d) Frequenzverdreifachung - 45 - 7.2.5 NLO-Effekte 3. Ordnung (kubische NLO-Effekte) Frequenzverdreifachung, Selbstfokusierung, Quadratischer Kerr-Effekt, quadratischer StarkEffekt, EFISH (Electric Field Induced Second Harmonic), Cotton-Mouton, Raman-, Brillouin-, Rayligh-Streuung Auftreten auch bei zentrosymmetrisch orientierten Materialien → niedrigster nichtverschwindender NLO Term → 3. Ordnung • Nichtlinearer (quadratischer) elektrooptischer Effekt → Kerreffekt: n ∝ E 2 • Selbstfokussierung: • Selbstphasenmodulation: Zeitabhängige Frequenz eines Lichtimpulses • Vierwellenmischung • EFISH: E = Eint + Eω → 2. NLO → Term ∝ E int Eω2 D. h. EFISH ist eine Nichtlinearität 3. Ordnung getrieben vom internen elektrischen Feld. - 46 - 8. Piezo- und Pyroelektrizität 8.1 Piezoelektrischer (elektromechanischer) Effekt In allen Festkörpern verursacht ein anliegendes elektrisches Feld E eine Polarisation P , die eine mechanische Deformation (z. B. eine mechanische Dehnung ε ) zur Folge hat. Diesen Effekt bezeichnet man als Elektrostriktion. Er zeigt eine quadratische Abhängigkeit: E2 ∝ P2 ∝ ε Elektrostriktionskoeffizient: g ijkl = ∂ 2 β ij ∂ 2 S kl = ∂Pi ∂Pj ∂Tkl Der Effekt der Elektrostriktion ist nicht umkehrbar. In kristallinen Werkstoffen, deren Kristallgitter ein Symmetriezentrum (zentrosymmetrisch) aufweist, tritt ausschließlich dieser Effekt auf. In kristallinen Werkstoffen, deren Kristallgitter kein Symmetriezentrum (nicht zentrosymmetrisch) aufweist, wird zusätzlich zum Effekt der Elektrostriktion eine lineare Abhängigkeit zwischen E , P und ε , die Piezoelektrizität, beobachtet. Dieser Effekt ist umkehrbar. Man spricht vom direkten piezoelektrischen Effekt, wenn die mechanische Deformation zu einer elektrischen Polarisation führt: ε ∝ P ∝ E , und vom reziproken (inversen) piezoelektrischen Effekt, wenn ein elektrisches Feld im Werkstoff eine Verformung verursacht: E ∝ P ∝ ε . Keine Piezoelektrizität: Ladungsschwerpunkte verschieben sich nicht gegeneinander. Polarisation in positiver y-Richtung: Piezoelektrizität! ∂Di ∂S jk 1 ∂Q ∂Q = = = ∂T jk ∂Ei A ∂p ∂F p : mechanischer Druck, F : mechanische Kraft, x : Probendicke, V : elektrische Spannung Piezoelektrischer Koeffizient: d ijk = V = ∂x ∂V Relevant für piezoelektrische Sensoren: d 33 − Koeffizient - 47 - F , Q = PA Bei Piezoelektrika kann der quadratische Effekt der Elektrostriktion gegenüber dem linearen Effekt der Piezoelektrizität vernachlässigt werden: u ij = d ijk E k + g ijkl E k E l → Piezoelektrizität ist Elektrostriktion getrieben von der Polarisation! Durch den Piezoeffekt und die Elektrostriktion kann also eine Umformung mechanischer Spannung in elektrische Signale erfolgen und umgekehrt. In der Voigt-Notation wird sowohl die mechanische Spannung als auch die mechanische Verzerrung durch Vektoren Tα und S α dargestellt mit jeweils 6 Komponenten. Der dreistufige Tensor d kann dann als Matrix dargestellt werden, wobei der zweite Index jk zu α zusammengefasst ist und jeweils von 1 bis 6 durchläuft. Beispiele: • Piezoelektrischer Tensor für Quarz: d iα • 0 = 0 d 31 Keramiken: d iα d11 = 0 0 0 0 0 d15 0 d 31 0 d 33 d15 0 0 0 − d11 0 0 0 d14 0 0 0 0 0 − d14 0 0 − d11 0 0 0 0 8.1.1 Anwendungen • Direkter Piezoeffekt („elektromechanische Kopplung“): Tonabnehmer, Mikrofone, Beschleunigungsmesser • Elektrostriktion: Ultraschallerzeuger - 48 - k2 k1 kovalent k1 - + H F H H H H H F H F F H H F F H H F F H F F F F F F F F H F F H H F F H H F F H F F 8.1.2 Modell: Piezoelektrizität in semikristallinen Polymeren (z. B. PVDF) k2 + b k1 - van-der-Waals a „Steife Kette“: Kovalente Bindung „Weiche Kette“: van-der-Waals Bindung Piezoelektrizität als Zusammenspiel von van-der-Waals und kovalenter Bindung in polaren Ketten. Piezoelektrischer Koeffizient: d = • nq k 2 − k1 a 2 k 2 + k1 Polymere: Dipoldichte-Piezoelektrizität (dimensionale Piezoelektrizität): Verformung - 49 - d 33 < 0 • Kristalle: Intrinsische Piezoelektrizität: Verformung 8.1.3 Piezoelektrische Sensoren Ersatzschaltbild: Ladungsmessung mit Ladungsverstärker: Beschleunigungssensoren: - 50 - d 33 > 0 8.2 Pyroelektrischer (elektrothermischer) Effekt Spontane Polarisation: Von spontaner Polarisation wird gesprochen, wenn in einem Material eine Polarisation vorliegt, ohne dass äußerliche Einflüsse dafür verantwortlich sind. Sie ist bedingt durch die Struktur und kann nur in Materialklassen mit einer einzigartigen polaren Richtung auftreten. Somit scheiden alle Materialien aus, die ein Symmetriezentrum besitzen. Der pyroelektrische Effekt wird durch eine Temperaturabhängigkeit der spontanen Polarisation hervorgerufen. Eine Temperaturänderung von 1 K kann eine Änderung des (von der Polarisation verursachten) elektrischen Feldes von bis zu 105 V/m (Maximalwerte extremer Stoffe) hervorrufen. Damit eignen sich einige pyroelektrische Kristalle hervorragend zur Verwendung in thermoelektrischen Messgeräten. Häufige Anwendung finden sie auch in Detektoren für Wärmestrahlung. Der pyroelektrische Effekt kann durch den piezoelektrischen Effekt überlagert werden, da durch Temperaturschwankungen Volumenänderungen auftreten, die wiederum eine mechanische Spannung nach sich ziehen. Man spricht dann von sekundärem pyroelektrischen Effekt. Änderung der spontanen Polarisation eines Materials auf Grund einer Temperaturänderung (d. h. eine Ladungsantwort auf Grund einer Temperaturänderung): pi = ∂Di ∂Ξ 1 ∂Q = = , Q = PA , Ξ : Entropie ∂T ∂Ei A ∂T 8.2.1 Beispiele für piezo- und pyroelektrische Materialien Pyroelektrische Werkstoffe sind auch stets piezoelektrisch! 8.2.2 Signalmessung - 51 - 9. Phasenübergänge 9.1 Der Glasübergang in amorphen Festkörpern Polymere und andere „weiche“ Materialien können als kristalline, teilkristalline, flüssigkristalline (orientierte) oder amorphe Festkörper vorliegen. In den meisten Fällen kann man durch schnelles Abkühlen aus der Schmelze ein amorphes Glas herstellen. Ein Glas ist thermodynamisch instabil, aber kinetisch stabil, d. h. die Kristallisation ist durch das Einfrieren der molekularen Bewegungen gehemmt. Die Temperatur, bei der aus der unterkühlten Schmelze das Einfrieren geschieht (ein Knick in der Enthalpiekurve), hängt von der Kühlrate ab, d. h. der Glasübergang ist eine Funktion der gewählten Zeitskala. Wenn ein Glas sich mit der Zeit an die Enthalpie des Kristalls annähert, spricht man von Alterung. 9.1.1 Theorie des freien Volumens (Cohen & Turnbull) Mittleres freies Volumen: V = Ve + V f (T ) Ve : van-der-Waals Eigenvolumen V f (T ) : intramolekulares freies Volumen V f (T ) = Vα (T − Tr ) , α : lineare thermische Ausdehnungskoeffizient Tr : Referenztemperatur Dolittle-Gleichung (entspricht VFTH-Gesetz): - 52 - Ve Vα (T − Tr ) η : Viskosität η ∝ exp 9.1.2 Theorie des freien Volumens (Eisenbach) WLF-Gleichung: ln aT = − C1 (T − Tr ) C 2 + T − Tr Doolittle-Gleichung: ln η = ln A + 1 1 α (T − Tr ) ln aT = B − → ln aT = B f f f + α ( T − T ) r r r B und f r : Freie-Volumen-Parameter (WLF-Parameter) f = f r + α (T − Tr ) mit B , f : anteiliges freies Volumen f und 9.1.3 Gegenüberstellung thermischer Übergänge 1. Phasenübergang 1. Ordnung: Schmelzen der kristallinen Bereiche 2. Phasenübergang 2. Ordnung: Intramolekulare kooperative Effekte G. Glasübergang Glasübergang ist kein thermodynamischer Gleichgewichtszustand – Übergang vom ergodischen ( T > TG ) zum nicht-ergodischen ( T < TG bzw. T = TG ) Zustand. - 53 - 9.2 Ferroelektrizität Als Ferroelektrika bezeichnet man eine Klasse von Materialien, die auch ohne äußeres angelegtes elektrisches Feld eine spontane Polarisation PS (Orientierungspolarisation) besitzen (analog zum Ferromagneten). Diese heißt remanente Polarisation Pr . Die Eigenschaft der Ferroelektrizität verschwindet oberhalb einer charakteristischen Temperatur, der Curie-Temperatur TC . Diesen Übergang bezeichnet man als Phasenübergang. Oberhalb dieser Temperatur verschwindet die Ordnung, d. h. es findet ein Übergang vom Ferroelektrikum zum paraelektrischen Werkstoff statt. Hier überwiegt die der Ordnung entgegen wirkende thermische Energie. Für Paraelektrika gilt die lineare Proportionalität zwischen der Polarisation und dem elektrischen Feld. Wenn unterhalb der Curie-Temperatur das angelegte elektrische Feld variiert und gegen die Polarisation aufgetragen wird, erhält man eine Hysteresekurve. Ordnungsparameter: x o a2 a2 x a1 x a1 v v o: n1a1 + n2 a 2 1 v 1 v x: n1 + n1 a1 + n 2 + n2 a 2 2 2 Freie Energie: o v v o: n1a1 + n2 a 2 1 v 1 v x: n1 + n1 a1 + n 2 + + η n 2 a 2 2 2 η : Ordnungsparameter 1 F ' ' (T , p )η 2 + ... → Symmetrie → 2 F (T , p,η ) = F (T , p ) + A(T , p )η 2 + B (T , p )η 4 + ... F (T , p,η ) = F (T , p ) + F ' (T , p )η + - 54 - 9.2.1 Strukturelle Phasenübergänge Es existieren 32 Symmetrieklassen für Kristalle mit den Symmetriesystemen triklin, tetragonal, hexagonal, monoklin, orthorombisch, trigonal und kubisch. Von diesen besitzen 11 ein Symmetriezentrum und haben daher keine polaren Eigenschaften. Von den restlichen sind 20 piezoelektrisch, d. h. sie zeigen eine elektrische Polarität unter äußerem Druck bzw. eine Dehnung durch Anlegen einer äußeren elektrischen Spannung. Eine ausgezeichnete polare Achse haben 10 dieser Klassen. Sie besitzen eine spontane Polarisation PS (= Dipolmoment pro Volumeneinheit). Diese ist im Allgemeinen temperaturabhängig, d. h., man beobachtet einen Ladungstransport von oder zu der Oberfläche eines Kristalls aufgrund einer Änderung der Temperatur. Daher bezeichnet man solche Systeme als pyroelektrisch. Die Ursache für die spontane Polarisation sind die nicht deckungsgleichen Ladungsschwerpunkte von positiven und negativen Ionen in solch einem Kristall. • Stabilitäts-Kriterium: bei Temperatur T ist diejenige Struktur stabil für welche die freie Energie F minimal ist. • Verschiebungsübergang: nicht-polar → polar; (geladene) Untergitter verschieben sich (z. B. Kristalle, Keramiken) • Ordnung-Unordnung-Übergang: Ausrichten (lokaler) elektrischer Dipolmomente (z. B. ferroelektrische Polymere) • Die 10 Symmetrieklassen: • 1 2 m mm2 3 3m 6 6mm 4 4mm Info – Einteilung von Festkörpern: Einheitszelle (Basis) 7 Kristallsysteme (Raumgitter) (triklin, monoklin, hexagonal, trigonal, orthorhombisch, tetragonal, kubisch) 14 Bravaisgitter 32 Symmetrie (Kristall)-Klassen - 55 - Kristallstruktur (fcc, bcc, hcp, sc, Eisgitter, Flussspatgitter, Diamantgitter, Kalkspatgitter) 9.2.2 Eigentliche Ferroelektrika – Phänomenologische Beschreibung der Ferroelektrizität (Landau-Devonshire Theorie): Ordnungsparameter → Polarisation → Dielektrische Verschiebung F = F0 + αD 2 + γ 4 D4 + δ 6 D 6 + ... Devonshire-Näherung: α = A(T − T0 ) , T0 : Phasenübergang 1. Ordnung: = TC bei Abkühlung (thermische Hysterese) Phasenübergang 2. Ordnung: = TC bei Abkühlung und Erwärmung E= • ∂F ∂D T Phasenübergang 1. Ordnung: Sprunghafte Änderung struktureller Art (Sprung des Ordnungsparameters), Latente Wärme, PS springt unmittelbar auf endlichen Wert unterhalb TC . γ < 0 , δ > 0 : E = αD + γD 3 + δD 5 E = 0 → D = PS → 5 Lösungen für F Für T > TC folgt Curie- Weiss-Gesetz mit Curie-Temperatur Thermische Hysterese! T0 < TC TC = T0 + → β > T0 4αγ Phasenübergangstemperatur auf TC − T0 beschränkt - 56 - • Phasenübergang 2. Ordnung: Stetige Änderung, keine Latente Wärme, PS steigt stetig von Null sobald T unterhalb TC fällt. γ > 0 , δ = 0 : E = α D + γD 3 E = 0 → D = PS → 2 Lösungen für F : PS = 0 PS = für T > TC (paraelektrischer Zustand) A γ (TC − T ) für T < TC (ferroelektrischer Zustand) D = ε 0 εE → ε = 1 D 1 ∂D 1 1 = = ε 0 E ε 0 ∂E ε 0 ∂E / ∂D E = 0 → D = PS → 2 Lösungen für ε 0 ε : ε 0ε = ε 0ε = - 57 - 1 für T > TC A(T − TC ) (Curie-Weiss-Gesetz) 1 2 A(TC − T ) für T < TC • Elektrostriktion bei Ferroelektrika: F = F0 + αD 2 + T= γ 4 D4 + 1 D 6 + CS 2 − ξSD 2 , S : mechanische Verformung 6 2 δ ∂F ξ = CS − ξD 2 = 0 → S = D 2 → S ∝ D 2 ∂S C D = ε 0εE + PS → S ∝ ε 02 ε 2 E 2 + 2ε 0εEPS + PS2 Piezoelektrischer Effekt 9.2.3 Ferroelektrische Domänen In ferroelektrischen Materialien sind Dipolmomente lediglich innerhalb einzelner Domänen ausgerichtet, an der Domänengrenze ändert sich die Richtung der Polarisation. Uniachsiales System → nur 180°-Dömänen Die Breite der Domänen ist proportional zur Wurzel aus der Kristalldicke. Größere Domänen werden durch die depolarisierenden Felder unterdrückt und kleinere durch die ansteigende Domänenwandenergie (proportional zur Domänenwandfläche). Verringert man dagegen die Kristalldicke bis in die Größenordnung der Domänenwandbreite, kann die Depolarisationsenergie nicht länger durch die Bildung von Domänen minimiert werden. In diesem Fall wird die Ferroelektrizität des Systems zerstört. Diese untere Grenze ist sehr stark von der Kristallsymmetrie und -geometrie abhängig und ist z. B. Thema aktueller Forschung im Bereich moderner Speichertechnologien. 90°-Dömänen führen zu starken mechanischen Verspannungen. Domänengröße bei ferroelektrischen Polymeren: < 30 nm - 58 - 9.2.4 Semikristalline Hierarchie bei ferroelektrischen Polymeren Nach dem Polungsprozess: Festsitzende Ladungen zwischen den kristallinen und amorphen Bereichen → Kompensation des depolarisierenden elektrischen Feldes. Amorpher Bereich: Dielektrische Polarisation Kristalliner Bereich: Ferroelektrische Polarisation Polymerketten (kovalente bzw. van-der-Waals Bindung) (0.2 ÷ 0.8 nm) → Lamellen (20 ÷ 60 nm) → Kristallite (5 ÷ 50 µm) → Sphärolithe (50 ÷ 500 µm) → Polymerfilm 9.2.5 Relaxor Ferroelektrika Ein ferroelektrischer Relaxor ist durch folgende Eigenschaften charakterisiert: • Das Maximum der Permeabilität zeigt eine erhebliche Abhängigkeit von der Messfrequenz. Die sehr runden Maxima verschieben sich mit steigenden Frequenzen zu höheren Temperaturen und nehmen im Wert deutlich ab. Ein analoges Verhalten zeigen die Imaginärteile der Permeabilität, die mit den dielektrischen Verlusten gleichzusetzen sind. • Oberhalb des Phasenübergangs beobachtet man noch eine lokale Polarisation. Diese korrelierten Bereiche werden auch als Nanocluster bezeichnet. • Nach dem Abkühlen im Nullfeld findet man einen strukturellen Symmetriebruch in der polaren Phase. Eine makroskopische spontane Polarisation wird aufgrund von mikroskopischen Domänenstrukturen jedoch nicht beobachtet. Diese so genannten Nanodomänen entstehen beim Abkühlen am Phasenübergang aus den polaren Clustern und weisen unterhalb von TC eine extrem verlangsamte Dynamik auf. Man spricht in diesem Fall auch vom „Einfrieren“ der Domänen. Daraus erklärt sich auch, - 59 - dass die Hochtemperatur-Relaxor-Phase ergodisch ist, die polare Tieftemperaturphase jedoch nicht. 9.2.6 Anwendungen von Ferroelektrika und Materialbeispiele Ferroelektrika zeigen in der Regel große Werte der dielektrischen Funktion auf. Den Polarisationszustand kann man auch technisch nutzen in so genannten ferroelektrischen Datenspeichern (FeRAM). Der Vorteil gegenüber dynamischen RAMs ist die Nichtflüchtigkeit der gespeicherten Information. Da die spontane Polarisation sowohl Temperatur als auch druckabhängig ist, können Ferroelektrika sowohl zum Nachweis von Temperatur- (Pyroelektrizität) als auch Druckänderungen (Piezoelektrizität) verwendet werden. Der pyroelektrische Effekt wird vornehmlich in Infrarotsensoren zur Bewegungsmeldung eingesetzt, der piezoelektrische Effekt zur Funkenerzeugung in Feuerzeugen aber auch zur Ultraschallerzeugung in der zerstörungsfreien Werkstoffprüfung und Medizintechnik etc. Piezoelektrische Stellglieder werden für empfindliche Positioniereinrichtungen eingesetzt. Gefunden wurde der piezoelektrische Effekt bei einer gezielten Suche durch die Gebrüder Curie im vorletzten Jahrhundert (1880). Die ersten technischen Anwendungen wurden um 1918 diskutiert (Nicholson in den USA mit Mikrophonen, Lautsprechern und Langevin in Europa, Sonar). Piezoelektrika sind heute nicht mehr wegzudenken, Beispiele sind piezoelektrische Filter in der Fernseh- und Mobilfunktechnologie. In der Mobilfunktechnologie werden derzeit Filter mit Frequenzen im Bereich von 5 GHz entwickelt. • Entdeckung der Ferroelektrizität (Valasek, 1921) an einem Natrium-Kalium-Tartrat: Seignettesalz (1655 von E. Seignette synthetisiert, seit 1880 sind seine piezoelektrischen Eigenschaften bekannt (Echolote etc.), monokline Struktur ist ferroelektrisch) • KDP (Kalium-Dihydrogen-Phosphat) und dazu isomorphe Salze (Ursache Wasserstoff-Brücken-Bindung: H verschiebt sich aus symmetrischer Lage, Unordnungs-Ordnungs-Übergang) • Barium-Titanat (BaTiO3): Ferroelektrische „Modellsubstanz“ (1945, Vul und Goldmann, UdSSR; 1945, Hippel, Breckennidge, Chesley, Tisza, USA), Kristallstruktur: Perovskit (Verschiebungs-Übergang) • Ferroelektrische Polymere: Polyvinyliden-Fluorid (PVDF) und das Kopolymer mit Tri-Fluorethylen P(VDF-TrFE) (siehe „Spezielle Polymere“) - 60 - 10. NLO Polymere (Amorphe Polymere, Photonikpolymere) 10.1 Funktionspolymere Funktionspolymere haben sich als neuartige Materialien für hoch-technologische Anwendungen in Bereichen der Sensorik und Aktorik, optischen Datenspeicherung und Photonik erwiesen. Eine wichtige Klasse von Funktionsmaterialien sind Polymerelektrete, dielektrische Materialien mit einer quasipermanenten elektrischen Ladung. Polymerelektrete können in Ladungselektrete mit echten Ladungen und Dipolelektrete mit Polarisationsladungen auf Grund molekularer Dipole unterteilt werden. Diese Materialien haben wegen ihrer piezo- und pyroelektrischen bzw. nichtlinear optischen Eigenschaften Anwendungen in der Elektronik, Sensorik und Photonik gefunden. 10.2 Amorphe Polymere als Funktionspolymere Amorphe Polymere sind besonders für Photonikanwendungen wie optische Wellenleiter und Polymerfaser interessant. Sie sind transparent und zeigen diese Transparenz im Wellenlängenbereich der Telekommunikation. Man unterscheidet zwischen unpolaren bzw. schwach polaren Materialien für Isolierungen und passive Photonikelemente und stark polaren Polymeren für NLO Anwendungen 2. Ordnung. Die unpolaren Materialien zeigen kleine dielektrische Konstanten und werden daher auch als „low-k“-Polymere in der Halbleiterindustrie untersucht. Polare Photonikpolymere enthalten dipolare Moleküle mit einem konjugierten π-ElektronenSystem (Chromophore). Die Funktionalität dieser Polymerklasse wird durch Polung erzielt, d. h. durch eine nicht-zentrosymmetrische, quasi-permanente Orientierung der Farbstoffmoleküle in der Polymermatrix. Eine alltägliche und technologisch wichtige Anwendung finden diese Materialien in optischen Datenspeichern und wieder beschreibbaren CD-ROMs. 10.2.1 Organische Chromophore Bevor organische Moleküle für NLO-Anwendungen in Betracht gezogen wurden, dominierten die anorganischen Kristalle wie Lithiumniobat oder Kaliumdihydrogenphospat zuerst die Forschung und dann den Markt. Organische Chromophore besitzen jedoch eine sehr hohe NLO-Effizienz, die sich aus den in den ungesättigten Verbindungen nur relativ schwach gebundenen π-Elektronen herleitet. Diese konjugationsfähigen Elektronen lassen sich leicht durch äußere Felder verschieben und sind wesentlich für die gute Polarisierbarkeit der Chromophore verantwortlich. Die gegenwärtige Untersuchung vieler der in den vergangenen 150 Jahren synthetisierten Farbstoffe im Hinblick auf ihre NLO-Effizienz sowie die Möglichkeit der gezielten Synthese haben die Verwendung organischer Farbstoffe in der nichtlinearen Optik gefördert. Als Folge davon wurde die Erforschung von StrukturEigenschafts-Beziehungen intensiviert, um NLOphore gezielt für Anwendungen konfektionieren zu können. Vor allem die Verbesserung der thermischen Stabilität ist ein Forschungsschwerpunkt. Mittlerweile sind NLOphore entwickelt worden, die zumindest kurzzeitig Temperaturen von etwa 350 °C ausgesetzt werden können. Die π-Brücke (meist bestehend aus aromatischen und/oder Polyenstrukturen) wird von Elektronendonor- und -Akzeptorgruppen flankiert, die in π-Konjugation miteinander stehen. - 61 - Dadurch wird dem elektronischen Grundzustand bereits ein Anteil an ladungsverschobener bzw. ladungsgetrennter mesomerer Grenzstruktur beigemischt. Die elektronische Anregung dieser Chromophore wird durch einen HOMO-LUMO-Übergang beschrieben und hat dabei ausgeprägten „charge-transfer“-Charakter. 10.2.2 Polymersysteme Möglichkeiten, wie die Chromophore mit der Polymermatrix verknüpft sein können: Gast-Wirts- Seitenketten- Hauptketten- Quervernetzte Polymere Große Dipolmomente mit starken Donator (D) und Akzeptor (A)-Gruppen durch Vergrößern der π-Bindungslänge. Beispiele: Azo-, Stilben-Farbstoffe • Gast-Wirts-Polymere: Chromophore in der Polymermatrix gelöst, eingeschränkte Löslichkeit der NLO-Moleküle, geringe thermische Stabilität der Dipolorientierung; Beispiele: PMMA, PC, PI • Seitenkettenpolymere: Chromophore kovalent an die Polymermatrix gebunden, hohe Dipoldichte, gute thermische Stabilität orientierter Dipole, Nachteil: Auftreten flüssigkristalliner Phasen durch mesogene Gruppen zwischen den Seitenketten; Beispiel: Poly(Styrol-Maleinsäure-anhydrid) mit dem Azo-Farbstoff Dispersionsrot 1 (P(S-MA)-DR1): * CH2 CH O CH CH C C n * Hauptkette backbone N O C3H6 Verbindungsglied linker N C2H5 CH3 N N dye Farbstoff NO2 P(S-MA)-DR1 Typ 9511 von SANDOZ Optoelectronics (Farbstoffanteil 93 mol-%, Tg = 137°C) - 62 - • Hauptkettenpolymere: NLO-Dipole innerhalb der Polymermatrix, Dipolorientierung und -Relaxation schwierig, flüssigkristalline Phasen möglich • (Quer-)Vernetzte Polymere: Vernetzung durch thermische oder photochemische Prozesse, Reduktion der Dipolbeweglichkeit (= Erhöhung von Tg ), sehr gute thermische Stabilität der Chromophore 10.2.3 Polung Es gibt eine Reihe von Methoden, eine nicht-zentrosymmetrische Ausrichtung (Polung) der Chromophore in NLO-Polymeren zu erzeugen: • • • • • • Thermische Polung Elektronenstrahlpolung Koronapolung Gas-unterstützte Polung Optische Polung Lichtinduzierte Polung Allen gemeinsam ist das Einfrieren der geschaffenen Polarisation im Glaszustand. Polungsmethoden, die unterhalb Tg durchgeführt werden können, nennt man „kalte“ Polung (z. B. Optische und Lichtinduzierte Polung). Diese Methoden nutzen die lokale Änderung des freien Volumens um die Chromophore während des Polungsprozesses. • Prinzip Thermische Polung: - 63 - Nach Anlegen einer Gleichspannung und Erwärmen der Polymermatrix auf Temperaturen ≥ Tg koppeln die molekularen Dipole mit dem elektrischen Feld und nehmen eine gerichtete Orientierung ein. Diese nicht-zentrosymmetrische Struktur wird dann durch Abkühlung (unter Aufrechterhaltung des elektrischen Feldes) auf Temperaturen deutlich unterhalb Tg konserviert. Da sich solcherart gepolte Polymere ohne ein externes elektrisches Feld nicht mehr im thermodynamischen Gleichgewicht befinden, wird in diesen Systemen eine starke Tendenz zur Orientierungsrelaxation beobachtet. Liegt die Arbeitstemperatur ausreichend weit unterhalb der Polungstemperatur, ist die Relaxation kinetisch soweit gehindert, dass längerfristig die NLO-Aktivität des Materials erhalten bleibt. Wird das Polymer anschließend vernetzt, lässt sich die Relaxation der Chromophore weiter vermindern. Problematisch dabei ist oft die chemische Stabilität der Chromophore unter den Bedingungen der Vernetzung. • Prinzip lichtinduzierte Polung: Beim lichtinduzierten oder photoinduzierten Polungsprozess (PIP, photoinduced poling), eingeführt 1992 von Sekkat und Dumont, wird die Beweglichkeit der Farbstoffmoleküle unterhalb Tg erhöht, indem das freie Volumen lokal vergrößert wird. Das geschieht bei der Bestrahlung der Polymere mit Licht mit Wellenlängen im Absorptionsbereich der Chromophore („Pumpen“), wodurch die Dipole zu einer reversiblen trans-cis-Konformationsänderung angeregt werden. Der Azo-Farbstoff wechselt bei der Anregung von einem trans-Zustand in einen metastabilen cis-Zustand – die Wahrscheinlichkeit dafür ist durch cos 2 Θ p gegeben ( Θ p : Winkel zwischen dem elektrischen Feldvektor des Pumplichtes und der Dipolachse). Parallel zum Lichtfeld ausgerichtete Dipole werden daher bevorzugt angeregt. Der cis-Zustand relaxiert thermisch mit oder ohne Rückorientierung in den trans-Zustand. Nach mehreren solchen Photoisomerisationszyklen richten sich schließlich die meisten Chromophore senkrecht zum Lichtfeldvektor aus. Durch die - 64 - Strukturänderung bedingte erhöhte Dipolbeweglichkeit (vergrößertes lokales freies Volumen) können die Farbstoffmoleküle im elektrischen Feld ausgerichtet werden. µE Debye: < cos Θ p >≈ 3k B T (Langevin Näherung) Vorteil: Polarisation in der Tiefenrichtung des Polymers kontrollierbar (strukturierte Polung, Herstellung von Bimorphen). - - - + + + + + + + + + + + - - - - + + - + + + - - + + - - + - + + - - + Vdc + - - - + - - - + - + - - + + - + - - + + Vdc + - - - - thermal poling above Tg photoinduced poling below Tg Nachteil: Geringe thermische Stabilität der Dipolorientierung verlangt hohe Anforderung an die Polymersynthese (z. B. Einführung mesogener Gruppen) bzw. an die Prozessdurchführung. Gepolte amorphe Polymere gehören zur C ∞ Symmetrieklasse von Materialien → Suszeptibilitätstensor 2. Ordnung: χ ( 2) • 0 = 0 χ ( 2) 31 0 χ 0 0 0 0 ( 2) 31 ( 2) 31 ( 2) 33 χ Unerwünschter Materials: χ 0 χ 31( 2) 0 0 Nebeneffekt 0 0 0 der Polung: → Erhöhte Lichtstreuung in Lichtleitern! - 65 - Oberflächendeformationen des 10.2.4 Phasenanpassung In anorganischen NLO-Materialien ist ein hoher EO-Koeffizient mit einer großen Dielektrizitätskonstante verknüpft. Dies führt in der Anwendung zu einem hohen Energieverbrauch und relativ geringen Schaltfrequenzen. Zur Abschätzung der Effizienz eines NLO-Materials gerade auch in der technischen Anwendung wird daher die so genannte figure of merit (FOM) angegeben. n3r , r : EO-Koeffizient FOM = ε ∆n ≈ 0.1 ÷ 0.2 für trans-cis-Übergang Die Effizienz der Frequenzverdoppelung bzw. der Frequenzmischung hängen davon ab, ob die von den einzelnen Oszillatoren des Dielektrikums abgestrahlten Teilwellen konstruktiv interferieren. Konstruktive Interferenz wird generell in dispersiven Medien mit frequenzabhängigem Brechungsindex nicht eintreten. Die Welle der verdoppelten Frequenz wird von der Einfallenden Welle kohärent erzeugt. Eilt die Erzeugende der Erzeugten voraus oder hinterher, so kommt es nicht zur vollen konstruktiven Interferenz. Durch geeignete Wahl des Einfallswinkels → Anpassung der Phasen → Effizienz der Frequenzverdopplung von nahezu 100% SHG: TE/TM Grundmode → TE/TM 2. Harmonische Störungen im Wellenleiter (Dicken-, Brechungsindexschwankungen) → Kopplung der Moden Grundwelle + 2. Harmonische → unterschiedliche Phasengeschwindigkeiten, da sie unterschiedliche Brechungsindizes sehen → Phasenfehlanpassung ∆ . ∆ = 0 → Phasenanpassung („phase matching“, PM) • Keine strukturierte Nichtlinearität: ∆ = 0 → I SHG ∝ L2 ∆ ≠ 0 → I SHG ∝ L2 + oszillierende Funktion 2π mit der Periode α = ∆ • Strukturierte Nichtlinearität (z. B. cos(αy ) ): bei ∆ = α → SHG effizient → I SHG ∝ L2 → QuasiphasenAnpassung (QPM) bei ∆ ≠ α → SHG nicht effizient → I SHG klein - 66 - - 67 - 11. Semikristalline (Ferroelektrische) Polymere Semikristalline Dipolelektrete enthalten polare Kristallite, eingebettet in einer polaren, amorphen Matrix. Die Kristallite in den meisten solcher Polymere sind ferroelektrisch. Typische Beispiele dieser technologisch wichtigen Materialien sind PVDF und P(VDF-TrFE). Ferroelektrische Polymere werden durch Polung für piezo- und pyroelektrische Anwendungen funktionalisiert. Im Gegensatz zu NLO-Polymeren zeigen semikristalline Dipolelektrete starke piezo- und pyroelektrische Effekte aber schwache NLO-Effekte. 11.1 Kristallstruktur und Ferroelektrizität 11.1.1 Chemische Struktur CH2 CH2 CF2 CH2 CF2 n PVDF CHF x y n y=1-x P(VDF-TrFE) • Für semikristallines PVDF gibt es mindestens fünf unterschiedliche kristalline Phasen ( α , β , γ , δ , ε ). • Kristallinität für PVDF: ca. 50% ÷ 60% • In der kristallinen Form: t − oder g + − Konformation • Stabilste kristalline Phase bei Raumtemperatur: unpolare α − Phase ( tg + tg − − Konformation) • unpolare α − Phase → polare, ferroelektrische β − Phase (nur t − Konformation) durch mechanisches Strecken • unpolare α − Phase → polare δ − Phase durch elektrisches Feld • δ − und ε − Phase sind die Polaren „Gegenstücke“ zur α − und γ − Phase • Kristallinität für P(VDF-TrFE): bis zu 80% - 68 - • TrFE-Anteil > 18% → P(VDF-TrFE) kristallisiert direkt in die polare β − Phase • Tg = −40°C für PVDF und P(VDF-TrFE) → Amorphe Matrix ist bei Raumtemperatur im Gummizustand 11.1.2 Phasendiagramm • 50 ÷ 80 mol% VDF Ferroelektrische Phase Anteil: • Über 80 mol% VDF Anteil: kein Phasenübergang, da die extrapolierte Curie-Temperatur TC von 205°C höher als der Schmelzpunkt ( Tm = 80°C) liegt • 60 ÷ 80 mol% VDF Anteil: sprunghafter Phasenübergang 1. Ordnung mit thermischer Hysterese (Grund: weite Größenverteilung der Kristallite) • 50 ÷ 60 mol% VDF Anteil: kontinuierlicher Phasenübergang 2. Ordnung • Unterhalb 50 mol% VDF Anteil: Übergang von einem hoch-Temperaturparaelektrischen in einen niedrig-Temperatur-antiferroelektrischen Zustand • Durch Elektronenstrahlen: Übergang vom ferroelektrischen in ein Relaxorferroelektrisches Verhalten 11.1.3 Nachweis von Ferroelektrizität Schalten der Polarisation in einem elektrischen Wechselfeld (Hysterese) → Sawyer-Tower Schaltung: CS Q µC = =D= 2 A A cm C S >> C F σ = Vout - 69 - 11.1.4 Anwendungen von ferroelektrischen Polymeren Zerstörungsfreie Dickenmessung: Hydrophone: Auflösung: 1 ÷ 2 µm Infrarotsensoren: - 70 - 12. Flüssigkristalline Polymere Flüssigkristalline Polymere (Liquid Crystalline Polymers, LCPs) sind hochmolekulare Stoffe, die einen flüssigen aber geordneten, anisotropen Zustand zeigen (anisotrope Fluide, mesomorphe Phase). Der flüssigkristalline Zustand ist neben dem amorphen und kristallinen Zustand als mögliche Festkörperzustände ein „Zwischenzustand“ hin zum flüssigen Zustand. Flüssigkristalle sind teilgeordnete Systeme, die in thermodynamisch stabilen Phasen vorliegen. Da sie zwischen der isotropen Flüssigkeit und dem Festkörper angesiedelt sind, spricht man auch vom 4. Aggregatzustand der Materie. Man unterscheidet ganz allgemein zwischen thermotropen und lyotropen Flüssigkristallen. Während bei ersteren der flüssigkristalline Zustand nur in einem bestimmten Temperaturbereich auftritt, wird die Ausbildung von flüssigkristallinen Phasen bei letzteren durch Wechselwirkungen mit verschiedenen Lösungsmitteln bedingt. 12.1 Strukturen und Unterteilungen Mesogene Materialien → Materialien, die Flüssigkristallinität hervorrufen können • Stabförmige Moleküle: • Scheibenförmige (diskotische) Moleküle: • Nadelförmige Moleküle • Amphiphile Moleküle - 71 - • Hauptketten LCPs: • Seitenketten LCPs: 12.2 Klassifikation mesomorpher (flüssigkristalliner) Phasen 12.2.1 Stabförmige Moleküle • Nematische Phase: • Cholesterische Phase: • Smektische Phase: Moleküle entlang einer Vorzugsrichtung orientiert ( n : Direktor) Chirale Moleküle → Direktor n beschreibt eine Helix Moleküle in Schichten gleicher Dicke angeordnet Smektik A (a): Orientierung senkrecht zur Schichtebene Smektik C (b): Orientierung mit Neigungswinkel θ zur Normalen - 72 - 12.2.2 Diskotische Moleküle • Nematische Phase: • Kolumnare Phase: 12.3 Anwendung – Flüssigkristall-Anzeigen Flüssigkristall-Anzeigen (Liquid Crystal Displays, LCDs) sind heute jedermann vertraut, sei es durch einfache monochrome, numerische Anzeigeelemente in Taschenrechnern und Armbanduhren, alphanumerische Anzeigen in Küchen- oder auch Messgeräten oder hochauflösende Farbdisplays in Laptop-Computern und Camcordern. 12.3.1 Nematische Flüssigkristalle Die nematische Phase ist diejenige, die in allen heute gebräuchlichen Anzeigeelementen verwendet wird. Auf Grund der dielektrischen Anisotropie und der elastischen Eigenschaften des nematischen Flüssigkristalls lassen sich dessen optische Eigenschaften zwischen gekreuzten Polarisatoren leicht durch Anlegen von elektrischen Feldern modifizieren, was in den TN- (twisted nematic) und STN-Zellen (supertwisted nematic) zur Informationsdarstellung genutzt wird. Dies sei kurz am Beispiel des Schaltens einer TNZelle veranschaulicht. Eine solche Zelle besteht aus zwei Glassubstraten, die jeweils mit einer transparenten ITO-Elektrode (Indium-Zinn-Oxid) und einer Orientierungsschicht (häufig geriebenes Polyamid) versehen sind. Die Substrate werden so angeordnet, dass sie einen Abstand von ca. 5–10 mm aufweisen, der mit dem Flüssigkristall gefüllt ist, wobei die untere gegen die obere Orientierungsrichtung um 90° gedreht ist. - 73 - Durch elastische Wechselwirkungen mit den begrenzenden Substraten bildet das Direktorfeld des Flüssigkristalles eine um 90° verdrillte Struktur aus. Das einfallende Licht wird durch den ersten Polarisator linear polarisiert, und die Polarisationsrichtung folgt im feldfreien Zustand (ohne angelegte Spannung) der Verdrillung. Das Licht kann nach Durchgang durch die Zelle einen gekreuzt stehenden Analysator passieren, wird durch einen Spiegel reflektiert und durchläuft auf gleichem Wege wiederum die Zelle, die somit hell erscheint. Legt man eine Spannung an, richten sich die Moleküle mit ihren Längsachsen entlang des elektrischen Feldvektors aus (bei positiver dielektrischer Anisotropie). Einfallendes linear polarisiertes Licht passiert unbeeinflusst die Zelle und wird vom gekreuzt stehenden Analysator absorbiert – die Zelle erscheint dunkel. Nach Abschalten der angelegten Spannung bildet der Flüssigkristall durch elastische Wechselwirkungen wiederum die verdrillte Struktur aus. Auf diesem Prinzip beruhen alle heute gängigen LCDs, von einfachen, unbeleuchteten Taschenrechneranzeigen bis zu den ungleich komplizierteren, rückwärtig beleuchteten Laptop-Displays: ein Einschaltvorgang, der dielektrisch getrieben ist, und ein Ausschaltvorgang, der auf elastischen Wechselwirkungen beruht. In dieser Funktionsweise finden sich auch die Nachteile der nematischen Anzeigen begründet. Die Umorientierung der optischen Achse (des Direktors) findet senkrecht zur Substratebene statt, was eine relativ starke Blickwinkelabhängigkeit bewirkt. Dies, obwohl bei Bankautomaten sicherlich wünschenswert, stört bei Computerbildschirmen eher. Dieses Problem wurde erst in der letzten Zeit durch den Einsatz der TFT-Technologie (thin film transitor) vermindert, bedingt allerdings auch deutlich erhöhte Produktionskosten. Ein weiterer Nachteil nematischer Displays liegt in den relativ langsamen Schaltzeiten, die im Bereich von ca. 100 ms (Taschenrechner) bis bestenfalls 30 ms (Laptops) liegen. Gängige Anzeigeelemente sind somit nicht voll video-tauglich, da hierfür Schaltzeiten kleiner 30 ms benötigt werden. Dabei handelt es sich wohlgemerkt um die Summe aus Ein- und Ausschaltzeit. Die Einschaltzeit, als dielektrischer Schaltprozess umgekehrt proportional zum Quadrat der elektrischen Feldstärke, lässt sich durch Vergrößerung der angelegten Spannung verringern. Die Ausschaltzeit, bedingt durch elastische Wechselwirkungen des Flüssigkristalls mit den Substraten, ist allerdings feldunabhängig. Ein großer Vorteil nematischer Anzeigen gegenüber solchen, die auf smektischen Phasen basieren, ist ihr „Selbstheilungseffekt“. Jeder hat sicherlich schon einmal auf den Bildschirm seines neuen Laptops gedrückt und war dann doch froh, als nach ein paar Sekunden die durch mechanische Umorientierung erzeugten bunten Farben (Doppelbrechung) wieder verschwanden. Oder, dass der Taschenrechner, nachdem er bei sommerlichen Temperaturen im Auto vergessen wurde (wobei der Flüssigkristall in die isotrope Phase übergegangen ist) nach Abkühlung wieder funktionierte. Vorteil gegenüber neuen Technologien ist sicherlich auch die Tatsache, dass es sich bei nematischen Displays mittlerweile um eine etablierte Technologie handelt, welche (relativ) preiswert produziert werden kann. 12.3.2 Ferroelektrische Flüssigkristalle (Ferroelectric Liquid Crystals, FLCs) Schon 1975 haben R. B. Meyer et al. durch Symmetriebetrachtungen gezeigt, dass alle smektischen Phasen mit Neigungswinkel θ , die aus chiralen Molekülen aufgebaut sind, eine spontane Polarisation PS aufweisen. Ist diese spontane Polarisation in einem elektrischen Feld zwischen zwei Richtungen schaltbar, so spricht man von ferroelektrischen Flüssigkristallen (in den meisten Fällen handelt es sich um die Smektisch C-Phase). Wie auch bei ferroelektrischen Festkörpern beobachtet man eine Hysterese, wobei in flüssigkristallinen Systemen die üblichen PS − Werte um einen Faktor 102 kleiner sind als bei Festkörpern. - 74 - Informationsdarstellung in Displays: Die FLC-Zelle wird so orientiert, dass die Richtung der optischen Achse in einer Domäne, z. B. + θ und + PS , entlang einer der Polarisatoren steht. Wird nun ein positives elektrisches Feld + E angelegt, so schaltet die andere Domäne von − PS nach + PS , was mit der Umorientierung der optischen Achse von − θ nach + θ verbunden ist. Einfallendes Licht erfährt keine Doppelbrechung und das gesamte Blickfeld erscheint dunkel. Wird nun die angelegte Spannung abgeschaltet, so bleibt im Gegensatz zu nematischen Anzeigen die Orientierung der optischen Achse des FLC erhalten, zeigt also einen Gedächtnis-Effekt. Erst bei Umkehrung des elektrischen Feldes nach − E schaltet die spontane Polarisation von + PS nach − PS , verbunden mit einer Umorientierung der optischen Achse von + θ nach − θ . Diese Umorientierung findet im Wesentlichen in der Substratebene statt. Einfallendes Licht erfährt nun eine Doppelbrechung und das Blickfeld erscheint hell. Das eigentliche, viel versprechende Einsatzgebiet ferroelektrischer Flüssigkristalle liegt in der Herstellung von video-tauglichen Flachbildschirmen. Die Schaltzeiten liegen im unteren msBereich, insbesondere weil im Gegensatz zu nematischen Displays der Ausschaltprozess nicht elastisch getrieben, sondern ebenso wie der Einschaltprozess durch das elektrische Feld bestimmt ist. Der oben angesprochene Gedächtnis-Effekt wirkt sich dabei positiv auf die Leistungsaufnahme des Displays aus, da wiederum im Gegensatz zu nematischen Anzeigen ein Pixel nur dann angesteuert werden muss, wenn es verändert werden soll. Auch die Blickwinkelabhängigkeit ist deutlich geringer, da der Schaltprozess, im Gegensatz zu konventionellen nematischen LCDs, im Wesentlichen in der Substratebene stattfindet. Ein Nachteil smektischer Phasen in Displays ist ihre relativ geringe Stabilität gegenüber mechanischen Deformationen. Anders als bei nematischen Anzeigen, die den oben angesprochenen „Selbstheilungseffekt“ zeigen, heilen Deformationen der smektischen Schichtstruktur üblicherweise nicht aus. Artikel zu LCD-Bildschirmen (inkl. TFT-Technologie) → Ch. Weber, „Nicht mehr in die Röhre gucken“, Physik Journal 3 (2004) Nr. 7, pp. 46 – 47 - 75 - 13. Biopolymere Biosysteme sind extrem entfernt von Gleichgewichtszuständen. Biosysteme entwickeln sich: • Festkörper, weiche Materie: Ensembleeigenschaften z. B. Sequenz der Monomere in einem statistischen Kopolymer • Biosysteme: Einzelsystem muss betrachtet werden z. B. nur wenige spezifische Monomersequenzen sind von der Natur in Proteinen ausgewählt Grundbaustein lebender Systeme ist die Zelle, die Materialien für Metabolismus und Reproduktion enthält, geschützt von der Umgebung durch eine Lipid-Doppelschicht. • Prokaryoten: z. B. Bakterien • Eukaryoten: Pflanzen, Tiere, einzellige Organismen, strukturierte Zellen aus membran-umschlossenen Organellen mit Gerüststruktur aus steifen, linearen Makromolekülaggregaten – „Cytoskeleton“ • Multizellulare Organismen beinhalten extra-zellulares Material 3 Klassen von Biopolymeren: Zucker, Nukleinsäuren, Proteine Zusätzlich: selbstorganisierte Membranen aus amphiphilen Molekülen 13.1 Die wichtigsten Bausteine 13.1.1 Zucker Zucker C(H2O)n dient z. B. als Energiespeicher (Stärke), Bausteine der Nukleinsäuren und als Bausteine in der Zellerkennung (Blutgruppen). Es gibt viele Isomere, die sich nur in der Orientierung ihrer Hydroxylgruppen unterscheiden: - 76 - Spiegelebene zwischen zwei Isomeren (gleiche Chemie) → rechtsdrehender (D) und linksdrehender (L) Zucker • Polysaccharide: Zuckerpolymere, in (Energiespeicher, strukturelle Elemente) • Glycoproteine: Polysaccharide „grafted“ an Proteine der Regel statistische Kopolymere 13.1.2 Fettsäuren und Lipide Lipide sind Hauptbestandteile von Zellmembranen. Fettsäuren dienen Energiespeicher. Sie bestehen aus einer Alkylkette und einer Carboxylgruppe. auch als • Gesättigte Fettsäuren: Alkylkette enthält nur Einfachindungen • Ungesättigte Fettsäure: Alkylkette enthält Doppelbindungen • Speziell: Phospholipide (Bestandteile der Zellmembran): polare (=hydrophile) Kopfgruppe, unpolare (=hydrophobe) Alkylkette 13.1.3 Nukleotide Träger der Erbinformation (DNA, RNA), Energielieferanten für fast alle molekularen Prozesse (ATP), Signalmoleküle → Nukleinsäuren: sequentielle Kopolymere aus vier Monomeren (Nukleotiden): • • DNA (Speichereinheit) RNA (Übertragungseinheit) Genetischer Code übersetzt die Information in der DNA - 77 - 13.1.4 Aminosäuren (Nukleinsäuren) Sequenz aus Nukleotiden. In der Natur gibt es 20 Aminosäuren, die sich alle nur in ihren Seitenketten unterscheiden. Dadurch werden sie entweder hydrophob / polar oder polar + negativ geladen oder polar + positiv geladen. Es gibt Isomerie, aber in Natur kommen nur L-Aminosäuren vor. 13.1.5 Proteine Proteine sind Kopolymere aus Nukleinsäuren. Sie dienen als Enzyme (Katalysatoren), strukturelle Proteine in der Zelle und im extrazellularen Material, Molekularmaschinen (z. B. Umwandlung von chemischer und mechanischer Energie für Stofftransport bzw. Beweglichkeit der Zelle) 13.2 Biologische Makromoleküle • Aufgebaut aus kleineren Moleküle (Anzahl relativ begrenzt) • Makromoleküle können Funktionen ausführen, die die einzelnen Baustein nicht vermögen • Diese Funktionen entstehen selbstorganisierten Struktur • Biologische Makromoleküle = Biopolymere • Beispiele: DNA Doppelhelix, Proteine wie Casein ( β − casein) aus einer hochkomplizierten - 78 - dreidimensionalen 13.2.1 DNA Doppelhelix Doppelhelix stabilisiert durch WasserstoffBrücken zwischen H und N2O DNA Beispiele: E. coli ( 4.7 ⋅ 10 6 Monomere / DNA, Länge 160 µm), Menschliche Zelle ( 280 ⋅ 10 6 Monomere / DNA, Länge 95 µm) 13.2.2 Proteinstrukturen • Primärstruktur (Sequenz der Aminosäuren): • Sekundärstruktur ( α − Helix, β − sheet): • Tertiärstruktur („Ball-and-Stick-Modell“): - 79 - 13.3 Organische dünne Filme • Langmuir Filme: organische Polymere auf flüssigen Oberflächen • • Langmuir-Blodgett Filme: organische Polymere auf festen Oberflächen Anwendungen: Sensoren, Detektoren, Displays, Komponenten in elektrischen Schaltkreisen • Baustein → Amphiphile Moleküle (z. B.: Carboxylsäuren und ihre Salze): polare Kopfgruppe (in Wasser löslich) und einem unpolaren Schwanz mit (CH2)n (n > 12) 13.4 Lipid-Doppelschichten • Aggregation der amphiphilen hydrophoben Effekt Monomere • Konfigurationsentropie der Wassermoleküle wird durch den direkten Kontakt mit der Kohlenwasserstoffkette reduziert • In der Doppelschicht → nur noch an den Enden Kontakt zwischen Wasser und Kohlenwasserstoffmolekülen - 80 - zu Lipid-Doppelschichten durch • Minimierung der Oberflächenenergie → temperaturabhängig, Volumen bleibt konstant) Vesikel (Liposom) (Form ist 13.5 Biomembrane • Grundbaustein für die räumliche Struktur von Biomaterie • Schematischer Aufbau: in Doppelschicht von Lipiden sind Proteine eingebettet • Membran nicht nur Stützwand der Zelle, sondern eine selektive Barriere für den Austausch zwischen Zellinneren und Umgebung (Membran verhält sich wie zweidimensionale Lösung für Proteine) • Aufgaben der membranständigen Proteine: Rezeptoren für Hormone, Drogen, Arzneimittel, Umweltgifte, Antikörper und Regulierung der Ionenpermeabilität der Membran • Beispiel: Eukariotische Zelle: - 81 - • Schematischer Aufbau von biologischem Materialtransport: - 82 - 14. Zellulare Materialien 14.1 Zweidimensionale zellulare Strukturen Festkörper mit isolierten Poren Zellularer Festkörper 14.2 Dreidimensionale zellulare Strukturen - 83 - 14.3 Metallische zellulare Materialien Nickel Kupfer 14.4 Keramische zellulare Materialien Zirkonat Mullit 14.5 Polymere zellulare Materialien Polyurethanschaum mit offenen Zellen Polyethylenschaum mit geschlossenen Zellen - 84 - 14.6 Natürliche zellulare Materialien Kork Balsaholz Korallen Tintenfischknochen Schwamm Hohlknochen Lilienblatt Pflanzenstiel 14.7 Zellulare Materialien als Lebensmittel Brot Meringue Schokoriegel Kartoffelchip 14.8 Fibrillare Materialien Filz Papier Baumwolle Fleece - 85 - 14.9 Anwendung zellularer Materialien • Thermische Isolation: Glas- und Polymerschäume • Verpackungsmaterialien: Polymerschäume (Polystyrol, Polyethylen, Polyurethan) • Baumaterialien: Holz, Knochen, künstliche Bienenwabenstrukturen • Auftriebsstrukturen im Schiffsbau • Weitere Anwendungen: Filter, wasserabweisende Textilien Träger für Tinten, Farben, Gleitmittel, 14.10 Strukturelle und mechanische Eigenschaften 14.10.1 Euler’sches Gesetz F: Zahl der Flächen E: Zahl der Kanten 2-dim.: F-E+V=1 V: Zahl der Ecken 3-dim.: -C+F-E+V=1 C: Zahl der Zellen - 86 - 14.10.2 Auxetische Materialien (negative Poisson-Zahl) Auxetische Netzwerke Konventionelle Bienenwaben-Netzwerke - 87 - 15. Komposite, Verbundmaterialien 15.1 Definition Materialien mit Eigenschaften, welche konventionelle Metalle, Keramiken oder Polymere nicht aufweisen. Beispiel - Flugzeugbau: Strukturelle Materialien mit geringer Dichte, mechanisch stabil, abriebs-, stoß- und korrosionsfest Metalllegierungen, Keramiken, Polymere können als „Komposite“ aufgefasst werden. Natürliche Kompositmaterialien: Holz, Knochen 15.2 Aufbauformen 15.3 Klassifikation verschiedener Komposite - 88 - 15.3.1 Zweiphasenkomposite Matrix und dispergierte Phase Einflussfaktoren: Konzentration (a), Größe (b), Form (c), Verteilung (d), Orientierung (e) 15.3.2 „Fiber-reinforced“ Komposite (a) kontinuierlich und ausgerichtet (b) diskontinuierlich und ausgerichtet (c) diskontinuierlich, nicht ausgerichtet 15.3.3 Polymermatrixkomposite (PMCs) Polymermatrix: üblicherweise vernetzende Epoxidharze 15.3.4 Metallmatrixkomposite (MMCs) Metallmatrix: leicht verformbare Metalle wie Aluminium, Magnesium, Titan, Kupfer 15.3.5 Keramikmatrixkomposite (CMCs) - 89 - 15.3.6 Strukturelle Komposite Sandwich-Panels Laminare Komposite 15.3.7 Weitere Kompositbeispiele • Komposite aus Polymeren und ferroelektrischen Keramiken für Ultraschall • Plastikmagnete • Polarisatoren, Nanokomposite (Halbleiternanopartikel) 15.4 Herstellung von Faserkompositen - 90 - 15.5 Physikalische Eigenschaften von Kompositen 15.5.1 Summengrößen • Physikalische Größe liegt zwischen den Größen der Matrix und der dispergierten Phase • Beispiel: dielektrische Funktion 15.5.2 Gemischte Größen • Zusammengesetzte physikalische Größen mit unterschiedlichem Mischungsverhalten • Beispiel: Schallgeschwindigkeit – abhängig von Dichte und mechanischer Steifigkeit 15.5.3 Produktgrößen Kombination unterschiedlicher physikalische Größen in der Matrix und dispergierten Phase, die zu neuen physikalischen Effekten im Komposit führen. - 91 -